Keywords
Diabetic foot ulceration, orthotic, in-shoe pressure, shear, digital health, medical device
Diabetic foot ulceration, orthotic, in-shoe pressure, shear, digital health, medical device
Diabetic foot ulcerations are a significant medical, economic, and social burden. The NHS spends approximately £1 billion per year in England on diabetic foot care1 with diabetic foot ulcer care being the largest factor, and reducing the prevalence of ulceration by one-third would equate to a saving of £ 250 million annually2. The occurrence of diabetic ulceration is high, with an estimated 10% of people with diabetes having an ulcer at some point in their life, diabetic ulcers preceding more than 80% of amputations in people with diabetes1, and over 50% of all major lower limb amputations performed between 2017/18 and 2019/20 were reported in patients with diabetes3. The mortality rates after major amputation due to diabetic foot ulcers are also alarmingly high4. Despite decades of clinical research and advances in our understanding of ulceration genesis, there remains a lack of practical solutions that empower patients to prevent diabetic foot ulcers through real-time monitoring of risk and behavioral interventions during daily activities5,6.
Existing approaches for diabetic foot ulcer risk management primarily focus on the classification of risk status (low, medium, high) to guide the provision of educational support for foot health self-management and frequency of clinical review7, or provision of offloading interventions8. This inevitably leads to an increased burden on the health service to provide managed foot care and creates challenges for adherence and effectiveness of interventions as a preventative approach.
Current NICE guidance and the national diabetic foot audit highlighted the need for effective prevention strategies and “intensive monitoring for people at risk of diabetic foot problems”1,9. Continuous monitoring has also been advocated as a means of delivering behavioral interventions in real-time and reducing ulceration risk in ways that are personalized to an individual’s risk status10,11. Measuring and managing pressures applied to the foot sole, a known risk factor for ulceration, are recommended for offloading interventions8. However, the available technologies and prior research have focused on the measurement of only compression pressures, and this neglects the important contribution of shear forces to ulceration genesis12,13. This results in poor sensitivity and specificity of risk predictions10,14.
Furthermore, these approaches are generally incapable of accounting for the dynamic and personalized nature of everyday activities, the nature and frequency of bouts of loading and unloading, and their long-term impact on foot tissue metabolism, and thereby, diabetic foot ulcer risk15. Activities such as walking, stair climbing, and prolonged standing all generate varying loading conditions on the foot16. These conditions change over time due to factors such as deformity and tissue exposure and are directly linked to lifestyle choices (e.g., occupation, domestic circumstances, leisure activities)10.
To address this pressing issue, we developed a Load Monitoring and Intervention System (LOMIS) that uses novel 3D force sensors to provide a real-time, comprehensive assessment of the five dimensions (5D) of load associated with diabetic foot ulcer risk: compression forces, two shear forces, and physical activity, all over time. By providing a more nuanced, real-time, and multi-dimensional approach to monitoring diabetic foot ulcer risk factors than has been possible before, this technology aims to fill a critical gap in the literature and practice, potentially revolutionizing the way we understand and manage diabetic foot care.
As a step toward downstream evaluation of clinical effectiveness, the primary aim of this study was to demonstrate the safety and functionality of LOMIS as a novel medical device for use by people with diabetes who are at risk of foot ulceration. A secondary aim was to establish the feasibility of examining the effectiveness of LOMIS within a larger-scale trial.
Key Objectives of the study are:
1. To evaluate the functionality of LOMIS in a daily living environment, we captured both the pressure and shear across a variety of activities.
2. To determine the safety of LOMIS for use independently by people with diabetes.
3. To establish the validity of LOMIS for measuring activities of daily living and the ability to identify and notify users of high-risk episodes.
4. To evaluate the suitability of LOMIS for independent use daily in participants own footwear. To assess feasibility of the LOMIS study recruitment and retention processes.
This is an early feasibility study to evaluate device compliance with regulations. This ensures that the device meets the required safety and performance standards when tested under conditions similar to the intended use.
The study was divided into three successive phases (Figure 1): Phase 1 was a laboratory study of LOMIS device use during controlled ambulation; Phase 2 was a pilot study of 5-day use at participants’ homes; and Phase 3 was a longitudinal study over 3 months of LOMIS use during free living. Between each phase, the trial steering committee agreed on progression as go or no-go, based on an evaluation of safety and functionality data. Alongside the evaluation of function, safety, and feasibility, Phase 3 will also explore the impact of LOMIS on users’ daily lives, including self-management and quality of life.
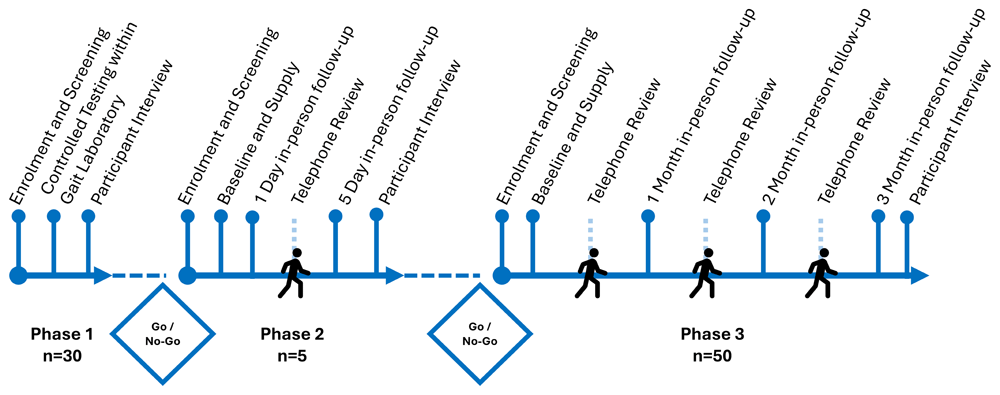
LOMIS Study timeline and progression, Phase 1 aimed to recruit 30 participants, Phase 2 aimed to recruit 5 participants, Phase 3 aimed to recruit 50 participants. Between each phase a Go-No-Go decision for study progression was made by the trial steering committee based on evidence collected for recruitment and device safety.
Trial registration: ISRCTN87061146, Registered on 06/10/2022
Protocol version: 9, 12/02/2024
Reporting of this safety and performance clinical trial protocol follows the protocol items in the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)17 with additional detail of the intervention in line with the Template for Intervention Description and Replication (TIDieR) checklist and guide18.
A Patient and Public Involvement and Engagement (PPIE) panel was established during the initial setup of the project to support the development of the LOMIS device and has played a key role in shaping the research question and designing the phases of the LOMIS study. The panel will meet quarterly throughout the trial, with a PPIE representative participating in both trial management group and steering committee meetings.
The PPIE panel’s responsibilities include contributing to the design and development of the LOMIS device, advising on how it can be applied to manage foot health, and monitoring the progress of the study. The panel will also provide guidance on recruitment strategies and patient-facing materials used in the study as well as participate in or support the dissemination of study findings. All PPIE activities were conducted in accordance with INVOLVE guidance19.
Phase 1 was conducted within the gait laboratory of the University of Salford, and the participants were provided with standard footwear. In Phases 2 and 3, the device was be fitted into the participants’ own footwear and used during typical daily activities. In Phases 2 and 3, device supply and review appointments took place either at the University of Salford or within NHS podiatry clinics at the North Manchester Diabetes Centre within the Manchester Foundation NHS Trust (MFT), community clinics within the Northern Care Alliance Foundation NHS Trust (NCA), and community clinics within the Cornwall Partnership Foundation NHS Trust (CFT).
To ensure the transferability of knowledge gained across the three phases, the same inclusion and exclusion criteria were applied to each. Participants must have a confirmed diagnosis of diabetes (Types 1 and 2) and feet classified as low, moderate, or high risk based on the International Working Group for the Diabetic Foot (IWGDF) risk stratification7. Participants must be capable of providing informed consent, aged between 18 and 85 years, and have self-reported capability to walk unaided and without stopping for 25m. Participants will be excluded if they are pregnant, have a body mass index of more than 50 kg/m², or have current participation in another clinical investigation of a medical device or a drug. Participants with dementia or uncorrected psychological impairment limiting compliance with the study were excluded. Given the safety focus of this study, to reduce the risk of harm, participants will be excluded if they have vascular complications and severe lower extremity artery disease, inner ear pathology or other serious underlying balance dysfunction, pathological conditions limiting the ability to walk unaided, severe retinopathy, or visual impairment that limits the normal use of smartphones (upon which the LOMIS device would rely). To minimize participant risk and the influence of comorbidities, participants with any of the following will be excluded: prior major injuries to the lower limb, active foot or leg ulceration, major amputation of the lower limb or foot, foot infection, active Charcot joint, foot deformity preventing orthotic provision, and currently using lower limb orthosis, which limits ankle movement or walking aids that offload the foot.
Participants continued to receive all usual concomitant care throughout the trial. Concurrent participation in other interventional studies was not permitted. Participants were advised that they should follow normal practice regarding foot care, and if they encountered problems related to their foot health requiring treatment during the trial, they were asked to stop using the LOMIS device and seek care urgently.
Participants were recruited through the podiatry clinics at each study site (MFT, NCA, and CFT). Potential participants were identified from clinical databases, clinic lists, or during routine clinical assessments by screening against inclusion criteria. Potential participants were provide a phase-specific participant information sheet (PIS)20. For Phase 1, potential participants may be enrolled on the same day; for Phases 2 and 3, potential participants were provided no less than seven days after providing the PIS to confirm interest. All potential participants were provided with an opportunity to ask questions pertinent to participation.
After confirming eligibility, a member of the study team confirmed that the participant understands the requirements of the study and the use of the novel device (explaining the intervention and highlighting any possible benefits or risks related to participation). Potential participants were given time to discuss the study and raise any questions that they may have. If the participant was willing to participate, they were asked to sign the consent form. No clinical trial procedures or device use were performed prior to consent. Fully informed written consent will be obtained by a member of the trial team in accordance with the good clinical practice guidelines.
No study has tested the LOMIS system in a home setting for patients with diabetes who are at risk of foot ulceration. As the first human trial, to establish safe use in line with medical device requirements for this population, a gradual progression of device exposure and appropriate oversight was required, and the sample size was determined on a pragmatic basis to allow effective monitoring and safety for users.
For Phase 1, a target of 30 participants, with 10 participants from each category of low, moderate, and high risk of diabetic foot ulceration, was proposed on a pragmatic basis and in line with similar within-lab orthotic intervention studies. For Phase 2, a target of five participants was selected to allow close monitoring of device use over a short period to evaluate usability and safety prior to progression to the longer-duration study. For Phase 3, a target of 50 participants was selected to allow for dropout over the study period and anticipate that 20 participants will complete the 3-month home use trial.
Development of the intervention. The LOMIS device is a functional prototype that has been developed through iterative bench and controlled testing. Iterations in PPIE activities contributed to user needs analysis and provided feedback on the requirements of the device throughout development. The initial technical development achieved within the LOMIS project following ethical and MHRA approval has been reported previously21,22 demonstrating initial safety, sensitivity, and reliability outcomes.
LOMIS device description. The LOMIS device is a load measuring, custom-made insole intended for use in patients with diabetes. Participants enrolled in all phases of the study will receive a pair of LOMIS devices for use in shoes, and will be advised to wear the device during all shod activities.
The LOMIS device is composed of a sensorized insole with embedded 3D load sensors, a hub with inertial measurement and data storage, and a flexible connector cable that allows the hub to be fitted to the outer collar of the footwear (Figure 2a). The system was provided to participants with a charger and required daily charging.
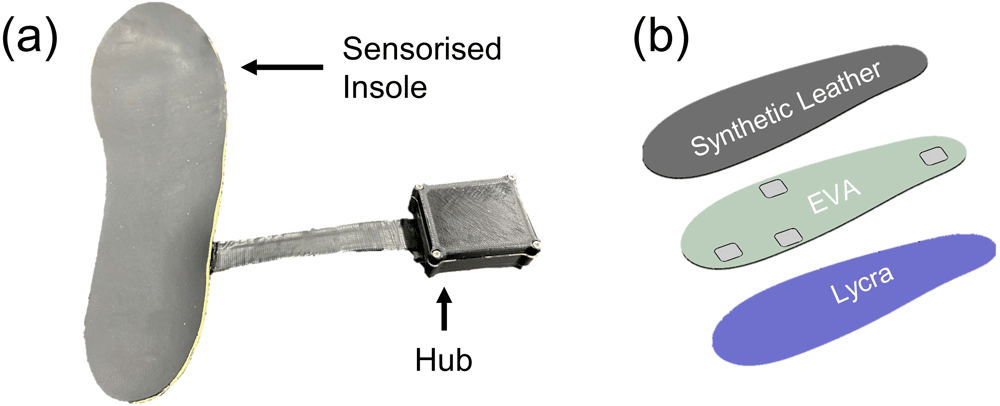
Image of the LOMIS Device showing (a) components including a sensorised insole, connector cable and hub. (b) layered structure of sensorised insole and positioning of sensor locations at the heel, 1st Metatarsal head, 5th Metatarsal head, and hallux.
The thin and flexible 3D load sensors were positioned at relevant load-bearing sites representing the heel, 1st Metatarsal head, 5th Metatarsal head, and hallux (Figure 2b). Each sensor had an active surface area of 20 mm × 20 mm, allowing for shoe movement and anatomical variation across individuals. The firmware within the LOMIS hub processes raw sensor data and stores the measured 3D load on an onboard SD card21.
Use within participants footwear and modification. LOMIS is permitted for use in footwear with a collar below the ankle, consistent with the clinical context for its eventual use, and may be used in combination with prescription orthotics and should be placed between the foot and orthotic. Modifications to the insole component can be made to ensure an optimal fit within the shoe, which includes trimming of the arch, toe, and heel regions.
Training in use of LOMIS. All clinical staff received training in the LOMIS system within the trial setup. Participants were trained in the use of LOMIS during supply appointments. A research team member checked the fit within the footwear and provide guidance about appropriate footwear for use with LOMIS. An instruction for the use of documents was provided for all participants at the point of supply.
This study will support the development of the LOMIS device in line with medical device requirements and inform the feasibility of a full trial of device effectiveness for the early identification of ulceration. The study phases were designed to progressively build evidence to support each outcome, while increasing exposure, user independence, and managing risk associated with the novel technology.
Primary outcome. The primary outcome was the functionality and usability of LOMIS in a daily free-living environment for the intended purpose of capturing plantar pressure and shear data during a range of daily activities.
Secondary outcomes. The secondary objectives can be divided into two areas: those addressing the device’s potential for use in the daily living environment, and those addressing the feasibility of a larger-scale trial and clinical implementation.
Safety, validity, and suitability for use in daily living. The safety of LOMIS is assessed by monitoring the frequency and nature of adverse events or device faults compared to the expected rates for patients at risk of diabetic ulceration.
Validity of LOMIS for the identification and classification of different physical activity and ulcer risk profiles (which would result in patient notification of ulceration risk) compared to recorded high-risk events (via adverse event reporting) and self-reported physical activity.
The suitability of LOMIS is assessed based on data describing usability, user experience, adherence, and integration into footwear. Engagement of participants as system users throughout the study (e.g., hours of use per day) will permit the adoption of a user-centered design approach.
Feasibility of LOMIS for larger scale study and clinical implementation. The feasibility of a future study to evaluate the device is assessed by monitoring the study recruitment, retention, intervention utilization, and participants’ compliance with LOMIS.
To estimate the potential effectiveness and standard deviations of a range of candidate outcome measures (physical activity, foot care activities, EQ5D, and MoBQoL) to establish an optimum way of evaluating changes in mobility and health-related quality of life.
Based on the analysis of LOMIS data collected from these studies, we also hope to establish an initial risk classification model by inputting pressure, shear, and physical activity data (over time) and clinician inputs.
Baseline assessment. Baseline data collection includes demographics (age, sex, marital status, ethnicity, education, occupation, and typical daily physical activity) and relevant medical history (diabetes type, time since diagnosis of diabetes, prior foot problems, fasting blood sugar, HbA1c, history of macro-and microvascular complications, end-stage renal disease, retinopathy, and any existing offloading footwear and orthotic provision) alongside the initial measurement of anthropomorphic characteristics (height, weight, body mass index), foot risk status (tactile sensory assessment, vibration sensitivity assessment, sensory symptoms assessment, peripheral arterial assessment, presence and severity of foot deformity), physical activity, quality of life (EQ5D and MobQol), and prior clinical service use (CSRI).
Controlled ambulatory activity. In Phase 1, participants undertook a series of controlled ambulatory tasks that reflect free-living physical activities but are done in the safety of a laboratory context. Participants wore appropriately sized standard therapeutic diabetic footwear (Omar 11, Fisio Duna).
Activities included level walking at self-selected, slow and fast speeds, incline/decline walking at self-selected, slow and fast speeds, stair climbing at self-selected, slow and fast speeds, quiet standing for 10 min, sitting with feet on the floor (5 min), sitting with feet raised on a chair (5 min), and a combined sit-to-stand (5 repetitions).
Supply and fitting of LOMIS for Phase 2 and 3. Participants had LOMIS fitted into their existing off-the-shelf (i.e., retail) or custom-made footwear (depending on the participants’ individual podiatric requirements). Each participant was fitted with a single LOMIS insole on the right-hand side. An equivalent left-hand side insole without sensors is also provided to be used concurrently as a pair to ensure an equal fit within both shoes. LOMIS devices accommodating different foot sizes ranging from 5 to 11 feet were provided to the participants accordingly. The LOMIS insole may be slightly modified by trimming it to fit the individual’s own footwear, where necessary. Training on the use of the LOMIS device was provided at the point of supply and supported through an instruction for use guide.
In-person follow-up. For Phase 2, a follow-up assessment were carried out at the University of Salford on day 1 (24 h after supply) and day 5. For Phase 3, follow-up assessments were carried out at either the University of Salford or within the NHS site on months 1, 2, and 3 after supply (Figure 1).
At each in-person follow-up, the participants’ feet were investigated for any new signs of redness, callus, blistering, ulceration, or other damage to the plantar surface. Follow-up assessments will include the following: adverse event monitoring, device fault logging, foot risk assessment (tactile sensorial assessment, vibration sensitivity assessment, sensory symptoms assessment, peripheral arterial assessment, presence of foot deformities, passive ankle dorsiflexion, and first MTP ROM), clinical service receipt, quality of life measures, physical activity monitoring, and device usability assessment. The LOMIS device will be replaced, and all device data for the prior use period was downloaded.
Tele-review and monitoring and retention. Telereviews were conducted by either a member of the research team at the University of Salford or within the NHS site by telephone and will occur at regular intervals between in-person assessments (Figure 1). These reviews provide an opportunity to identify potential harm early and ensure that participants continue to complete study documents and follow the advice provided on device use. Each telereview identified any issues, adverse events, or faults with device use; a short summary of daily physical activity was captured; and a mobility and device questionnaire was completed.
Criteria for discontinuing withdrawal of intervention. Participants who are unable to continue within the study due to adverse health conditions or events were withdrawn from device use, and the device was recovered. Participants may withdraw from the study or device use at any point. For participants who have stopped device use, the option of continuing within the study was given to allow the assessment of feasibility outcomes.
A series of clinical assessments are conducted at each in-person follow-up by an HCPC registered podiatrist or orthotist to determine the current risk of foot ulceration. The IWGDF risk stratification system was used to classify participants into low-, moderate-, and high-risk categories7.
Tactile sensory assessment. The Semmes–Weinstein monofilament examination is conducted to evaluate tactile sensorial deficits in four weight-bearing areas of each foot (plantar face of the hallux and first, third, and fifth metatarsal heads) according to practical guidelines on the management and prevention of diabetic foot7.
Vibration sensitivity assessment. Vibration sensitivity is assessed using a tuning fork (128 Hz) perpendicular to the dorsal region of the distal hallux phalanx, at constant pressure. The participant was asked to report the point at which the vibration was diminished below perception, and the evaluator recorded the time until they could no longer detect the vibration in their hand. Values less than 10s are normal, greater than 10s is decreased vibration sensitivity, and if the participant does not perceive the vibration imposed by the tuning fork, it is classified as absent vibratory sensitivity23.
Sensory symptoms assessment. Sensory symptoms are measured using the NTSS-6 questionnaire, which quantifies the frequency and intensity of aching pain, burning pain, lancinating pain, prickling sensation, numbness, and allodynia in the participants' feet and legs24.
Peripheral arterial assessment. Peripheral arterial disease is determined by palpating the dorsalis pedis and posterior tibial pulses25.
The presence of foot deformities. The presence of foot deformities is determined through visual inspection, and the presence of only one deformity will be necessary for classification of deformed feet. The inspections determine the presence of small muscle wasting (interossei wasting sufficient to cause 'troughing' between tendons), hammer or claw toes, hallux valgus (determined using the Manchester scale), hallux limitus, bony prominences, prominent metatarsal heads, and Charcot arthropathy25–28. Examinations were also conducted to identify calluses and score callus severity at 12 distinct plantar sites per foot (first–fifth toes, first–fifth prominent metatarsal heads, midfoot, and heel). Scores will be no callus (zero), mild callus (1), medium callus (2), or severe callus (3). The total callus severity scores were recorded for each foot plantar calluses25.
Plantar pressure assessment. To assess whether the LOMIS device does not pose an increased risk to the participant, an in-shoe pressure assessment was conducted by comparing the peak pressure with LOMIS to the peak pressure with the participants’ standard insole. During level walking and at a comfortable pace, plantar pressures beneath the foot was measured concurrently using XSENSOR (Foot and Gait v4, XSENSOR® Technology Corporation, Calgary, AB, Canada) following a standard 12 step protocol for the assessment of diabetic foot29.
Clinical service use. Participants were interviewed using the Clinical Service Receipt Inventory (CSRI) form to create a record of clinical appointments related to foot health and diabetes care that have taken place since the previous review appointment. Where participants are not able to provide details, NHS researchers may also complete or confirm this through a review of the patient’s health record.
Quality of life. Participants completed two validated questionnaires to establish their perceived quality of life upon entry into the study. The EQ-5D-5L evaluates general health status30 while the focus of the MobQol is on the evaluation of mobility-related quality of life31.
Monitoring of physical activity. Physical activity was recorded using a written log for each participant divided into morning, afternoon, and evening for each day of device use. This measure was developed in collaboration with the LOMIS PPIE group to ensure an appropriate balance between the manageable request of participants and a detailed record of daily activities.
Device usability. Participants completed the Quebec User Evaluation of Satisfaction with Assistive Technology (QUEST)32 which includes domains assessing: user acceptance, Ease of use, Accessibility, Technical affinity of the user group, Levels of ‘Anxiety,’ Levels of ‘Interest,’ Compliance with intervention and Satisfaction with system.
Device faults. The reported and identified faults were recorded using a written log for each participant, which stratifies faults into component-affected (hub and insole) and severity (harm, data loss, and no data loss).
Participant interview on usability and suitability. To prioritize timely feedback on device usability and suitability, participants were invited to participate in optional interviews following the completion of their involvement in each study phase. The aim was to better understand their opinions of LOMIS and its intended use from a participant's perspective, for the purpose of rapidly impacting technical device development (i.e., not for the purposes of thematic analysis of user experiences). Interviews were selected to allow for adaptive questioning recognizing the early stage the insole system is at in its development and evaluation, and existing measures (e.g., questionnaires) may not capture all relevant experiences. The interviewer will take notes and rapid reports to inform the ongoing development and subsequent implementation of the LOMIS device.
Data management. All data was recorded to standardized data entry forms and within a paper case report form. Data recorded by NHS sites was stored on a password-protected NHS computer, or in the case of paper records, in a lockable filing cabinet. Paper documents from each site were digitized and uploaded to a secure online drive for transfer to the University of Salford.
Confidentiality. All participants were assigned a unique participant number (UPN). Each NHS site maintained a screening and recruitment log onto which the UPN will be recorded alongside the recruitment date and NHS number or an appropriate clinical reference number used at the NHS Site. This allows NHS staff to quickly identify the clinical records and treating clinicians for participants if needed and enables a rapid response in the case of Adverse Events. Details for participants who were initially identified but who were screened as ineligible for participation will be retained for the duration of participant recruitment to ensure that they are not subsequently included in the screening. The data for these participants will be destroyed after the completion of the study.
Members of the research team at the University of Salford may need to contact participants for routine follow-up, device maintenance, and support. To enable this, participants were informed of the need to share identifiable personal information (name, address, email address, and telephone number) collected by members of the NHS staff with the University of Salford research team and provide informed consent. Additionally, demographic personal details will be stored by the University of Salford to establish the study demographics, which will include age, sex, marital status, education, and occupation.
Personal details will be stored separately in the main study documents in a password-protected database on a University of Salford laptop, which is accessible only to members of the research team. Personal details will be stored by each NHS site and by the University of Salford research team for a maximum of three years following the end of the study, after which point the data will be securely destroyed.
Research data. All research data will be anonymized (labelled using UPN only) and recorded in Clinical Record Forms and paper documents (screening checklists and questionnaires). The research data included no identifiable personal information. Research data will be retained for up to 10 years and may be shared with project partners at the University of Southampton, University of Keele, and University of Bangor, as well as used for publication and dissemination of research findings. All publicly accessible data will be presented in anonymized form only.
The measures will be analyzed using descriptive statistical approaches. We present quantitative data with summary statistics and 95% confidence intervals as appropriate.
The primary outcome of device functionality in a daily living environment is assessed across all the three phases. The ability to capture pressure and shear will be assessed for a given range of known activities within Phase 1 and in the real-world setting (Phases 2 and 3) through measures of data capture failure rate (%sensors/devices not operational, %activity/use not operational), daily physical activity, device use (% shod daily physical activity with/without LOMIS), and durability (frequency of replacement/maintenance).
Safety analysis is assessed across all studies and will review the rates of adverse events (primarily the number of ulcerations), rates of device faults that affect the risk of harm, incidence of incorrect or inappropriate use identified through monitoring reviews, and engagement with clinical services (CSRI). Safety for in-shoe application will be assessed by comparing in-shoe plantar pressure with and without LOMIS.
The validity of the activity classification is assessed against self-reported physical activity logs and the number of missed/mislabeled physical activities. The validity of risk labelling for free-living data (Phases 2 and 3) is evaluated by comparing high-risk events (incidence of high shear, pressure, acceleration, and incidence duration) to the normal range for physical activities observed in Phase 1.
Suitability, usability, and user experience is assessed using validated usability questionnaires (QUEST), structured qualitative interviews, and monitoring of device faults/incidence of user error or misuse. Adherence is assessed through interviews and monitoring of LOMIS device use compared with self-reported typical daily activities. Integration into footwear is assessed by monitoring the number and type of footwear worn in combination with LOMIS. This will help identify key difficulties and deficiencies in the current device design or delivery, which can be clustered, prioritized, and rectified in response to user-identified needs.
Feasibility analysis. To assess the potential population that could receive LOMIS as an intervention, the number of potential participants eligible, approached, consented, and supplied in total and per site level will be reported. The number of participants who attended each study appointment and who continued to use the LOMIS device for the duration of the study will be reported, along with the number of complete monitoring and self-recorded measures (physical activity log). Compliance with LOMIS will be assessed through monitoring of recorded daily physical activity against estimated daily activity at baseline, with a target adherence of 80% LOMIS use during shod activities.
To explore the potential effects or interactions of LOMIS within the target population, the following were assessed for Phase 3: The effect on participation in activity will be assessed by monitoring physical activity type, duration, and intensity over extended use. The effect on foot care was assessed through clinical service engagement and structured qualitative interviews. The effect on quality-of-life measures will be assessed using the 5Q-5D-5L and MobQoL-validated questionnaires.
Initial risk metric models developed using real-world data will also be assessed against clinically defined risk status.
The trial management group consisted of the Chief Investigator (who isin charge of the study), the device manufacturer (who is responsible for reporting adverse events to the MHRA), the trial manager (who is in charge of the day-to-day management of the study), the study’s grant co-applicants, and the Principal Investigators or delegated person at sites delivering the intervention. Regular meetings are held according to the trial needs.
The study sponsor is the University of Salford, who provide research governance and clinical trial indemnity. The chief investigator is a member of the research team from the host institution and is involved in study design, collection, management, analysis and interpretation of the data, writing of reports and decisions to submit for publication.
A combined Trial Steering Committee (TSC) and Data Monitoring Ethics Committee (DMEC) were formed for this study because of the low number of participants anticipated. The LOMIS TSC/DMEC will comprise an independent chair, at least two independent members (including one member of the patient’s public involvement and engagement panel), 1–2 PIs (From NHS Sites), The Chief Investigator; Clinical Lead, and Device Manufacturer. Other collaborators may also attend meetings. The role of the LOMIS TSC/DMEC was to provide advice, through the Chair, to the Sponsor, funder, and the consortium associated with the grant-funded research programme.
The LOMIS TSC/DMEC made decisions regarding study progression and reviewed all serious adverse events that are thought to be related to the intervention and are unexpected. The group met quarterly or more frequently if requested.
Data monitoring was conducted by study statisticians. The role of the members is to monitor the data and make recommendations to the TSC/DMEC regarding any ethical or safety issues regarding the study.
We consulted with the independent Trial Steering Committee before we start recruitment into the LOMIS Study 3 and at quarterly intervals during the study. They made recommendations as to whether the trial should continue in its current form, continue with minor amendments to improve site/participant recruitment or follow-up, or continue with major amendments/close. The progression criteria were as follows.
(1) Device risk
Red = Serious device fault or serious device related adverse event
Amber = Multiple device related adverse events.
Green = 1–2 device related adverse events.
(2) Device faults during LOMIS Study 2:
Red = Serious device fault.
Amber = frequent (daily) per participant.
Green = 1–2 (total) per participant.
(3) LOMIS Study retention
Red = <50% attending in-person follow-up assessment.
Amber = 50–79% attending in-person follow-up assessment.
Green = ≥80% attending in-person follow-up assessment.
Surveillance for adverse events is conducted for the duration in which participants are involved in the LOMIS study. This is the responsibility of the chief investigator (CI) and Trial Manager, who will ensure that adverse events are documented and reported accurately. All study staff are trained in the requirements and procedures for reporting the events.
This study was designed to ensure that any adverse effects were treated urgently by the participants’ healthcare professionals. New ulceration and other critical clinical episodes (e.g., redness, blistering, or damage to the weight-bearing surfaces of the foot) were reported immediately to the Trial Manager. In the first instance, this will occur primarily through normal NHS services (contacting their foot clinic directly or A&E if out of hours). During follow-up, any participant identified as having had an adverse event or having stopped device use will be asked to attend an additional research clinic appointment to review if it is suitable to continue use of the device or to retrieve the device where continued use is not possible.
A Serious Adverse Event (SAE) is defined as any untoward occurrence that results in death, is life threatening, requires hospitalization or prolongation of existing hospitalization, results in persistent or significant disability or incapacity, consists of a congenital anomaly or birth defect, or is otherwise considered medically significant by the investigator.
This study records details of any SAEs that must be reported to the Research Ethics Committee (REC) and Medicines and Healthcare Regulatory Authority (MHRA). SAEs are only recorded and reported if the event is suspected to be related to an aspect of the research procedures (e.g., wearing the orthotic, completion of follow-up questionnaires, participation in qualitative interviews), which is an unexpected occurrence. Related and unexpected SAEs will be reported to the main REC and MHRA within 15 days of CI becoming aware of the event. In the present study,
The occurrence of adverse events during the trial was monitored independently by TSC and DMEC. The TSC/DMEC immediately saw all SAEs thought to be intervention-related, and saw summary of all SAEs not thought to be treatment-related by the Trial Management Group at the next scheduled meeting.
Expected adverse events and expected side effects. The expected adverse events and side effects of treatment that are related to the intervention include aches, pains, unusual feelings, and discomfort in the lower limb or other parts of the body as a result of altered biomechanics, or as a result of undertaking a new exercise regimen, new callus/corn formation, blisters, ulcers, skin irritation/injury including pressure sores, soft tissue injury, heat foot, tight shoes, feeling unstable, and falls. It is very unlikely (although not impossible) that death may occur, for example, following an accident from wearing orthoses. The expected side effects of the intervention will be recorded in the participant follow-up questionnaires. Participants were asked if they sought treatment for any problems caused by wearing the insoles or undertaking activity, and if treatment had been sought, whether the problems had fully resolved.
The results of the LOMIS study will be disseminated through open-access, peer-reviewed journal publications, conference presentations, diabetes networks, and health professional networks. All participants and the participating NHS sites will be informed of the study outcomes. The study’s findings will be disseminated to the public via social media platforms, project research engagement, showcase events, and the study website (www.lomis.co.uk).
Ethics approval was obtained from the University of Salford Ethics Committee (ID: 2236 – date of approval: 31/08/2021) and via IRAS (ID: 298091) and the West Midlands – Edgbaston NHS Research Ethics Committee (REC: 21/WM/0239 – date of HRA approval: 21/12/2021). This study is a registered clinical investigation in accordance with the UK Medical Devices Regulations 2002 and has been reviewed by the MHRA (CI/2021/0067/GB). This study was registered in the ISRCTN registry (ISRCTN87061146). The trial is conducted in compliance with the principles of the Declaration of Helsinki and good clinical practice. Protocol amendments have been approved by the REC and MHRA up to the current version of the protocol reported in this manuscript (V9, 12/02/2024).
The current management of diabetic foot does not effectively identify changes in ulceration risk nor enables patients to identify and respond to potentially harmful events as they occur in real time10. This insensitivity to day-to-day changes in risk factors is a risk factor. The LOMIS device offers opportunities for intensive real-time monitoring and multiple additional advantages over the current standard of care, which will be explored in this pilot and feasibility study.
The ability to measure shear and pressure in shoes will provide a more comprehensive understanding of the loads experienced by at-risk feet and may better inform clinical management12,13. This is strongly inferred from prior studies that show that the measurement of compressive pressure alone is a poor predictor of foot ulcer risk14. Within other clinical presentations of pressure-related tissue breakdown, the influence of shear pressure is also well evidenced33 which has allowed for the development of clinical strategies for managing both shear and pressure34. Furthermore, quantifying the typical daily exposure to pressure and shear forces during activity will add to our understanding of how clinical risk and varied cumulative stresses interact.
The emphasis on the measurement of daily activity within this study reflects the need to develop interventions that reflect and respect the personalized patient activity and lifestyle profiles of those at risk of ulceration35. This is also in line with current NHS strategies for personalized care and supported self-management36 allowing interventions and approaches that are more tailored to individual patient needs. Future approaches may include behavioral strategies linked to data from intensive monitoring that have been advocated11.
The adoption of digital healthcare technologies within the NHS requires a large amount of evidence to demonstrate effectiveness, safety, and cost benefits37 alongside regulatory approvals necessary to place a device on the market. The world health organisation also recommends robust monitoring and evaluation of all digital health interventions throughout development and when implemented in practice38. This high bar for technology implementation serves to protect end users, but also presents considerable challenges for the development and deployment of novel and disruptive technologies at a pace that can match the changes within the population (e.g., increased ulceration prevalence) and health services (e.g., staff shortage).
The progressive staged approach outlined in this study provides evidence to support the development of a novel medical device alongside the evaluation of clinical feasibility for larger-scale investigation and implementation. This pragmatic trial has been designed and adapted to address the needs of patients and clinical services to reduce barriers to downstream implementation at scale.
The outcomes of this study will evaluate the function, safety, suitability, and feasibility of LOMIS and inform the design and management of a larger study to investigate the efficacy and cost-effectiveness of the intervention and prevention of ulceration incidence.
Closed to recruitment.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)