Keywords
Paediatrics, infant, urinary tract infection, antibiotics
Paediatrics, infant, urinary tract infection, antibiotics
 How to cite: Waterfield T, McMullan R, Lyttle MD et al. Empirical oral AntibioticS for possible UTI in well appearing Young febrile infants (EASY) [version 2; peer review: 2 approved, 1 approved with reservations]. NIHR Open Res 2026, 5:104 (https://doi.org/10.3310/nihropenres.14103.2)
First published: 17 Oct 2025, 5:104 (https://doi.org/10.3310/nihropenres.14103.1)
Latest published: 12 Feb 2026, 5:104 (https://doi.org/10.3310/nihropenres.14103.2)
How to cite: Waterfield T, McMullan R, Lyttle MD et al. Empirical oral AntibioticS for possible UTI in well appearing Young febrile infants (EASY) [version 2; peer review: 2 approved, 1 approved with reservations]. NIHR Open Res 2026, 5:104 (https://doi.org/10.3310/nihropenres.14103.2)
First published: 17 Oct 2025, 5:104 (https://doi.org/10.3310/nihropenres.14103.1)
Latest published: 12 Feb 2026, 5:104 (https://doi.org/10.3310/nihropenres.14103.2)
We have revised the manuscript to improve clarity and transparency in response to reviewer feedback. Specifically, we have clarified international variation in guideline recommendations regarding the route of antibiotic therapy in young infants with suspected UTI and explicitly highlighted this ongoing debate as a key rationale for the trial. We have provided additional explanation for the inclusion of infants with caregiver-reported fever, noting its clinical relevance while acknowledging that planned sensitivity analyses were not feasible due to early trial termination following the pilot phase. We have clarified the pragmatic approach to urine collection, reflecting UK NICE guidance and real-world practice, and explicitly defined the day-28 secondary outcome as the requirement for additional parenteral antibiotic therapy to avoid ambiguity.
See the authors' detailed response to the review by Ghassan Ghssein
See the authors' detailed response to the review by Kazuki Iio
Urinary tract infections (UTIs) are the most common serious bacterial infection (SBI) in febrile infants, accounting for over 90% of all SBIs in this age group1–7. Diagnosing UTIs in young infants is particularly challenging due to several factors. Clinical features are often non-specific, such as poor feeding, irritability, fever, and vomiting. Urine collection is difficult, as infants are unable to provide a clean-catch midstream sample8–12.
These diagnostic challenges necessitate a cautious clinical approach. Current UK national guidelines recommend empirical treatment with broad-spectrum parenteral antibiotics for all febrile infants under three months of age with suspected UTI, pending culture results13,14. In contrast, for infants aged over three months, oral antibiotics are considered appropriate while awaiting culture confirmation15,16. This discrepancy in treatment recommendations stems from concerns regarding the absorption and efficacy of oral antibiotics in younger infants17.
However, substantial evidence indicates that, beyond the neonatal period (>28 days), oral antibiotics are well absorbed, well tolerated, and effective in treating UTIs17–21. The prevailing cautious approach contributes to significantly higher healthcare resource use in febrile infants compared to older children22. Reducing the use of “just in case” parenteral antibiotics could lower healthcare costs, alleviate pressure on paediatric inpatient services, and enhance patient and family experience by shortening hospital stays and minimising invasive procedures.
Two previous randomised trials have evaluated the use of oral antibiotics in this population. The first, conducted by Hoberman et al. in the United States and published in 1999, included 306 infants with febrile UTI, 144 of whom were under six months of age20. Infants were randomised to receive either oral or parenteral antibiotics. The study found no significant differences between groups in time to defervescence, microbiological cure, or complications such as recurrent UTI and renal scarring. Notably, treatment costs were significantly lower in the oral antibiotic group (US$1,473 vs. US$3,577)20.
Similarly, Montini et al. conducted a non-inferiority trial in Italy in 2007, comparing oral and parenteral antibiotics in infants with febrile UTI, including 186 infants under six months of age21. Their findings mirrored those of Hoberman et al., with no significant differences in clinical outcomes, including time to defervescence, microbiological cure, normalisation of inflammatory markers, or incidence of renal scarring.
Reflecting this evidence, several international consensus guidelines recommend oral antibiotics as first-line therapy for infants with suspected UTI; however, recommendations are not uniform. For example, the American Academy of Pediatrics supports oral antibiotic treatment in infants older than 28 days, whereas Swiss consensus guidance recommends oral therapy in infants over 60 days of age. These discrepancies highlight ongoing clinical uncertainty regarding the optimal route of antibiotic administration in this population and provide a key rationale for conducting this randomised controlled trial2,10,23.
The overall aim of the EASY trial was to assess whether oral antibiotics, given while awaiting urine culture results, are as effective as parenteral antibiotics. Effectiveness was measured by treatment failure, defined as the need for additional parental antibiotics and various secondary outcomes. The trial also aimed to evaluate the impact of oral antibiotics on healthcare resource use, costs, and selected outcomes through a cost-consequence analysis.
The EASY internal pilot aimed to assess the feasibility of recruiting a sufficient number of eligible participants to support progression to a definitive trial. The pilot evaluated recruitment rates, screening and consent procedures, and randomisation processes.
Patients, carers, and members of the public were involved from the outset of the EASY study. A PPI group of 12 individuals including parents with lived experience of UTIs in infants, met five times to inform study design. Additional input was gathered from children and young people. PPI members helped shape the research focus on antimicrobial stewardship and reducing unnecessary hospital stays, and contributed to defining inclusion criteria, consent timing, and safety measures for early discharge. They supported randomisation and helped refine the non-inferiority margin to 3–5%. Two secondary outcomes; time to defervescence and time to normal feeding were proposed by the group. A parent representative was a co-applicant and part of the Trial Management Group. The PPI group contributed to protocol development, participant materials, recruitment strategies.
Multicentre, randomised controlled, open-label, non-inferiority trial with embedded internal pilot, comparing parenteral antibiotics versus oral antibiotics for the management of suspected UTI in low-risk infants.
During the pilot phase of the EASY study, two key protocol amendments were implemented to improve recruitment feasibility and better align with clinical practice:
1. Modification of Inclusion & Exclusion Criteria:
The original protocol required participants to meet specific laboratory thresholds for C-reactive protein (CRP) and white cell count (WCC). Also, the original protocol excluded infants ‘requiring re-admission to hospital after birth for >24 hours’. The CRP and WCC criteria were removed and the exclusion criteria changed to ‘re-admission to hospital after birth for >24 hours for parenteral antibiotics.’ This was done to broaden eligibility and reduce unnecessary exclusions, based on feedback from recruiting sites and early screening data.
2. Adjustment to Timing of Randomisation:
Initially, randomisation was required before the administration of a second dose of parenteral antibiotics. This was revised to allow randomisation within 24 hours of hospital arrival.
These changes were made in consultation with trial oversight committees, participating sites and the trial management group, and were intended to enhance recruitment while maintaining the safety and scientific validity of the study.
Recruitment for the EASY trial was conducted across 21 paediatric emergency departments (EDs) and assessment units throughout the United Kingdom. Participating sites were drawn from the Paediatric Emergency Research in the UK and Ireland (PERUKI) network and the General and Adolescent Paediatric Research in the United Kingdom and Ireland (GAPRUKI) network.
Participants were screened from attendances to paediatric EDs and assessment units at recruiting sites. All individuals meeting the study’s inclusion criteria were entered into a screening log. For those not recruited, the reason for non-enrolment was documented.
Eligibility was assessed by a physician listed on the site’s Delegation Log, in accordance with the trial’s inclusion and exclusion criteria. Medical care and clinical decisions for trial participants were provided by appropriately qualified treating physicians.
Informed written consent was obtained from the parent or guardian of each participant by a trained member of the clinical team prior to inclusion in the trial. Families were provided with a participant information leaflet, consent form, and verbal explanation of the study. Consent was documented before randomisation and trial procedures. Participants could withdraw at any time without detriment, and data collected up to withdrawal were retained unless otherwise requested. General practitioners were notified of their patients’ participation via letter.
Participants were eligible for enrolment if all the following criteria were met:
Participants were excluded if any of the following criteria applied:
1. Gestational age at birth <30 weeks.
2. Discharge from hospital more than 7 days after birth.
3. History of re-admission to hospital that required treatment with parenteral antibiotics
4. Known or suspected structural renal abnormality.
5. Clinical evidence of sepsis and/or meningitis, including:
6. Receipt of vaccination within 48 hours before attendance.
7. Serum sodium <128 mmol/L (laboratory or blood gas sample).
8. Serum potassium >6.5 mmol/L (laboratory or blood gas sample).
9. Plasma creatinine >50 µmol/L.
10. Inability to tolerate oral medication.
11. Urine sample not sent for culture.
12. Received additional antibiotics (with the exception of the parenteral antibiotic administered within 24 hours of hospital attendance)
13. Declined consent for participation.
14. Not clinically well on global assessment
Initial clinical assessment and management of febrile infants under three months of age were conducted according to standard care protocols. Blood and urine testing were performed, and broad-spectrum parenteral antibiotics were administered as clinically indicated, in line with national guidelines and local policy. Urine can be collected by the method deemed most appropriate by the treating clinician. These initial interventions were not influenced by trial participation, and administration of parenteral antibiotics was not delayed for consent procedures.
Screening for trial eligibility was initiated once initial laboratory results were available. Only infants meeting the predefined “low-risk” criteria were invited to participate. Randomisation was required within 24 hours of presentation to hospital.
Eligible participants were randomised in a 1:1 ratio to one of two treatment groups:
Randomisation was performed using an automated web-based or telephone system employing randomly permuted blocks. Stratification was applied by recruitment site, sex, age group (29–60 days vs. 61–90 days), and prior antibiotic use. Allocation concealment was maintained through secure storage of the randomisation sequence in a restricted section of the Trial Master File, accessible only to the trial statistician.
Following informed consent, randomisation was completed by a trained and delegated member of the research team. Each participant was assigned a unique study number, which was used for all trial documentation and data collection. Enrolment into the study was documented in the participant’s medical record.
This was an open label, unblinded trial. Parents or guardians, healthcare providers, and outcome assessors were aware of the allocated intervention. This pragmatic design allowed for realistic evaluation of clinical effectiveness, including hospital admission decisions unrelated to antibiotic administration.
The trial statistician, who had no role in trial conduct, remained unblinded to facilitate data monitoring and linkage with the Data Monitoring and Ethics Committee (DMEC). The broader trial team was also unblinded to support data management, case review, and pharmacovigilance activities.
Both oral and parenteral antibiotics used in the EASY trial were administered in accordance with their licensed indications and standard clinical practice. As the safety profiles of these agents are well established, and their use did not exceed routine care, the trial was classified as a Type A Clinical Trial of an Investigational Medicinal Product (CTIMP). A risk-adapted approach to trial management was adopted.
The most prescribed oral antibiotics included cephalexin, co-amoxiclav, and trimethoprim. Parenteral antibiotics included ceftriaxone, cefotaxime, gentamicin, amoxicillin, cefuroxime, and co-amoxiclav. All study drugs were stored, prescribed, and dispensed according to local site procedures and manufacturer recommendations. No additional labelling or accountability measures were required beyond standard practice.
Antibiotic administration followed local prescription schedules. Treatment could be modified or discontinued based on clinical assessment or urine culture results. Adherence was monitored through inpatient prescription records and verbal reports from parents/guardians at follow-up. Use of concomitant medications, including antipyretics, was permitted and recorded.
The EASY trial was designed to evaluate the clinical effectiveness and cost consequences of oral versus parenteral antibiotics in febrile infants with suspected UTI. The primary clinical outcome of the full trial was the requirement for additional parenteral antibiotics within seven days of randomisation. A range of secondary outcomes were also planned, including requirement for additional parenteral antibiotics at day 28, time to recovery, adverse events, antibiotic adherence, quality of life, family impact, escalation of care at day 7 and day 28, length of stay and healthcare resource use.
The primary objective of the internal pilot was to assess feasibility, specifically the ability to recruit eligible participants to support progression to a full-scale trial. Feasibility outcomes included:
Number of sites opened during the pilot phase
Recruitment rate per site per month
Total number of participants recruited
Screening and consent process performance
Adherence to randomisation procedures
These metrics were evaluated against predefined stop-go criteria to determine whether continuation to the main trial was justified. The criteria were as follows:
The EASY trial was powered to detect non-inferiority in treatment failure rates between oral and parenteral antibiotics. Assuming a treatment failure rate of 0.5% and a non-inferiority margin of 2%, a total of 524 participants were required to achieve 90% power with a one-sided 97.5% confidence interval. Allowing for up to 10% loss to follow-up, the final sample size was set at 584 participants. The internal pilot phase was designed to recruit 99 participants, representing approximately 17% of the total sample size. This pilot was conducted to assess feasibility, including recruitment rates, site activation, and protocol adherence, rather than to evaluate clinical outcomes.
As this manuscript focuses on the internal pilot phase, analyses were limited to descriptive statistics to assess feasibility. Recruitment rates, site activation, and protocol adherence were summarised and compared against predefined stop-go thresholds. No formal hypothesis testing or clinical outcome comparisons were conducted. The full trial included intention-to-treat and per-protocol analyses to evaluate non-inferiority in treatment failure rates, alongside secondary clinical and health economic outcomes.
A total of 27 participants were recruited during the internal pilot (20 May 2024 to 13 March 2025) from 10 UK sites, representing 27% of the target sample size for the pilot (n=99). Of these, 14 (52%) were randomised to receive oral antibiotics and 13 (48%) to receive intravenous (IV) antibiotics. Following review of the internal pilot, the trial was terminated on 25 March 2025 due to poor recruitment. A flow diagram of recruitment is shown in Figure 1.
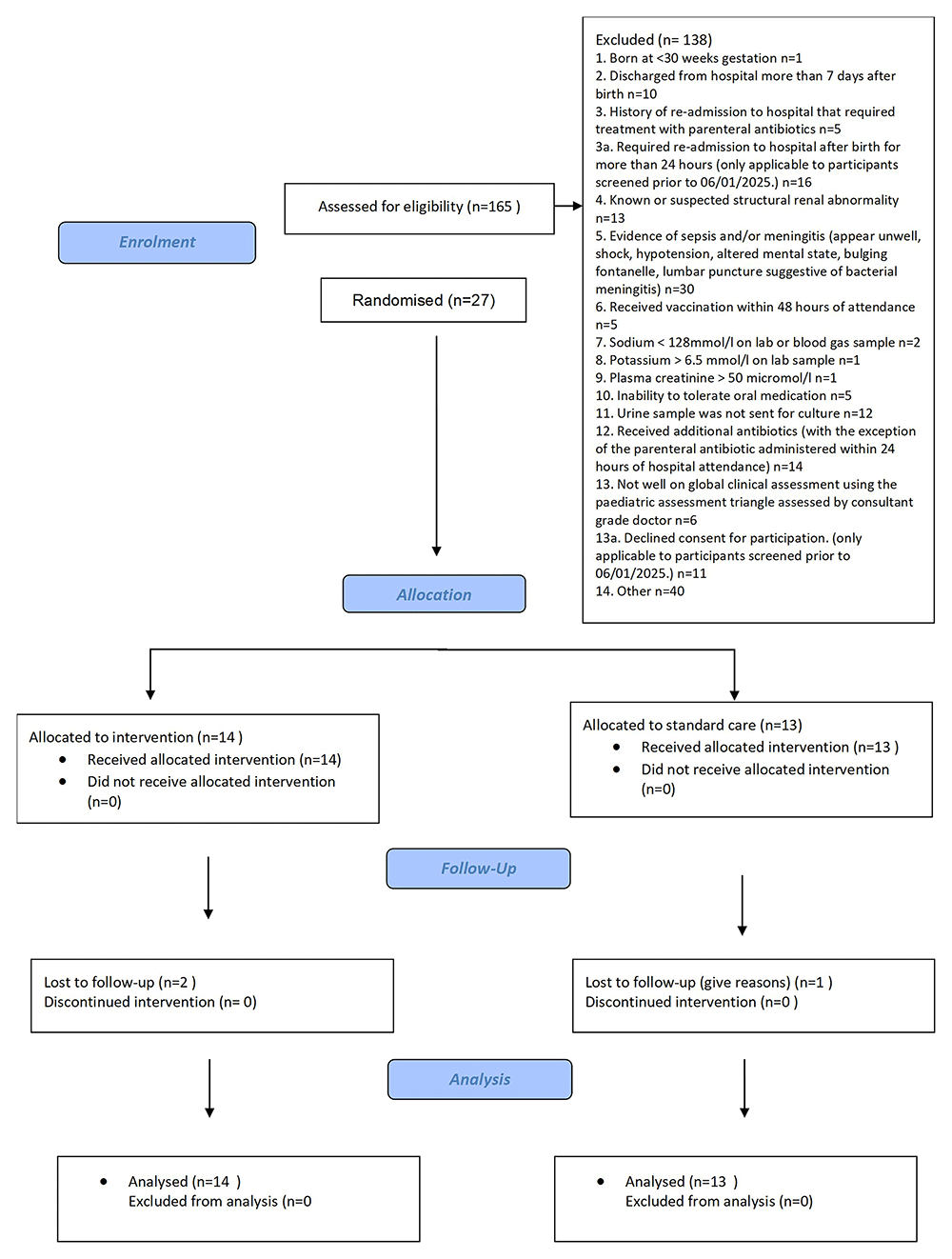
Participants in both groups were similar in age (mean ~57 days) the IV group included a higher proportion of females. Most participants were of White ethnicity, with limited representation from Asian, Black, and mixed ethnic backgrounds. Gestational age at birth was predominantly 39–40 weeks.
Vital signs and physical examination findings were broadly comparable across randomised groups, with minor differences in heart rate and respiratory rate. Urinalysis showed high positivity for leucocytes in both groups. Table 1 summarises the baseline characteristics.
Common presenting symptoms included decreased feeding and abnormal activity, which were more frequently reported in the oral group. Vomiting and diarrhoea were similarly distributed across both randomised groups. Blood results showed comparable inflammatory markers and electrolyte levels, although the IV group had slightly higher haemoglobin and platelet counts. Most participants received parenteral antibiotics prior to randomisation. These data are shown in Table 2.
Following randomisation, oral antibiotics were predominantly cefalexin (93%) and co-amoxiclav (7%), while IV antibiotics included amoxicillin, cefotaxime, or ceftriaxone (or combination of). Antibiotic treatment was discontinued before completion of the initially prescribed course in 43% of the oral group and 50% of the IV group, primarily due to negative urine culture results. Despite early discontinuation, adherence to prescribed doses was 100% in both groups. Table 3 summarises antibiotic administration and adherence.
Most participants completed the study as per protocol (86% oral, 92% IV). Protocol deviations occurred in both groups, with 11 events in the oral group and 7 in the IV group. The most common deviations were related to follow-up outside the scheduled timeline. No deviations were reported for consent procedures.
Treatment failure, defined as the need for additional parenteral antibiotics within seven days of randomisation, occurred in 3/14 (21.4%) participants in the oral group and 1/13 (7.7%) in the IV group. At day 28, no additional treatment failures were observed. No escalation in care; such as hospital admission, ICU transfer, change in antibiotic therapy, or death was reported in either group at day 7 or day 28. Time to defervescence was shorter in the oral group (mean 6.2 hours) compared to the IV group (mean 24.9 hours). Time to normal feeding and activity, as reported by parents, was slightly longer in the oral group, with high variability. Length of hospital stay was notably shorter in the oral group (mean 25.2 hours) compared to the IV group (mean 62.1hours). Clinical outcomes are summarised in Table 4.
Adverse events (AEs) were more frequent in the IV group (9 events observed in 5 patients) than in the oral group (3 events in 2 patients). Adverse reactions (ARs) and unexpected adverse reactions (UARs) were reported only in the IV group, 7 and 3 events respectively. Serious adverse events (SAEs) occurred in 1 patient in the oral group and 2 in the IV group. No serious adverse reactions (SARs) or serious unexpected adverse reactions (SUSARs) were reported.
Gastrointestinal disorders, renal and urinary tract infections, and skin disorders were the most reported AEs, with higher incidence in the IV group. ARs and UARs were exclusive to the IV group, primarily involving gastrointestinal and skin-related symptoms. Table 5 summarises the safety outcomes.
From initial presentation at the ED to hospital discharge, participants in the oral intervention group experienced shorter overall hospital stays, with a mean duration of 25.2 hours (SD 19.7) compared to a 62.1 hours (SD 46.1) in the IV group (mean 62.1) (Table 4). Inpatient admission rates were lower in the oral group (78.6%) versus IV (100%), with shorter inpatient durations (26.6 vs. 58.2 hours). Parents in the oral group reported less time missed from work (mean per participant 1.7 hours; 95% CI: -2.6–6.0) compared to the IV group (25.5 hours; 95% CI: -11.2–62.2). Time missed from usual activities was also lower in the oral group (30.0 hours; 95% CI -14.9 - 74.9) vs. 49.0 hours; 95% CI -71.8, 169.8).
The EASY trial was designed to assess the clinical and cost-effectiveness of oral versus IV antibiotics for febrile UTI amongst low-risk febrile infants. However, the trial was terminated following the internal pilot phase due to poor recruitment. While the internal pilot successfully demonstrated protocol adherence and safety across both treatment arms, several feasibility challenges emerged.
Recruitment during the EASY trial was significantly slower than anticipated, with only 27 participants enrolled across 10 sites during the internal pilot phase, well below the target of 99. This shortfall may reflect a lower-than-expected incidence of UTIs in the eligible population or a reluctance among paediatricians to randomise infants to oral therapy. Anecdotal feedback from sites suggests that clinicians often favoured IV antibiotics due to concerns about missing cases of bacteraemia, despite the study’s stringent low-risk inclusion criteria.
Indeed, two cases of bacteraemia were identified, one in each treatment group. Both these infants had an uncomplicated clinical course, with no escalation in care, supporting the safety of the trial protocol. Notably, no cases of meningitis were observed, further validating the risk stratification approach used to identify suitable candidates for oral therapy.
These challenges are common to many randomised trials that are terminated early due to poor recruitment24. A systematic review of 172 randomised trials found that the most common reasons for trial closure included an over estimation of disease prevalence and ‘prejudiced views of recruiters’ both of which affected the EASY pilot25.
Although recruitment was limited, preliminary findings suggest oral antibiotics may offer some benefits. Infants in the oral group tended to have shorter hospital stays and fewer inpatient admissions, with parents reporting less disruption to work and daily activities, although statistical significance testing was not undertaken on the data. These trends warrant further investigation in adequately powered studies. These findings align with previous trials demonstrating comparable clinical outcomes between oral and IV antibiotics in this population20,21.
However, recruitment data from the internal pilot phase indicate that continuing the trial would have been futile under the current design. The combination of slow recruitment, high exclusion rates, and clinician hesitancy indicates that substantial modifications would be required to achieve adequate sample sizes. Proposed adjustments such as increasing the non-inferiority margin and reducing the recruitment target may improve feasibility but risk undermining the trial’s statistical robustness and generalisability.
The internal pilot phase of the EASY trial has several limitations that should be acknowledged. First, the absence of an embedded qualitative or perspectives study limited our ability to fully explore and understand the barriers to recruitment. While anecdotal feedback from sites suggested clinician hesitancy and a preference for intravenous treatment, a formal evaluation of stakeholder views including those of parents and healthcare professionals would have provided valuable insights to inform future trial design and implementation strategies.
Second, the internal pilot study is underpowered to detect differences in clinical outcomes between the treatment groups. With only 27 participants recruited, the sample size was insufficient to draw definitive conclusions regarding treatment failure, safety, or efficacy. As such, any observed differences should be interpreted with caution.
This internal pilot phase of the EASY study demonstrated that while protocol adherence and safety were achievable, significant recruitment challenges driven by low disease prevalence and clinician reluctance made continuing the trial futile. These barriers, commonly reported in trials terminated early, underscore the importance of understanding recruiter perceptions and trial context. Future research should consider alternative designs or implementation strategies to evaluate oral antibiotic therapy in this population.
This study received ethical approval from the South Central – Hampshire A Research Ethics Committee on December 22, 2023 (Reference: 23/SC/0426).
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)