Keywords
Perinatal, platform, adaptive, preterm, impacted, fetal, respiratory, neurodevelopment
Perinatal, platform, adaptive, preterm, impacted, fetal, respiratory, neurodevelopment
This article describes the work undertaken to inform and set-up a complex platform trial, with a view to obtaining grant funding for a set of related research questions from which to launch the platform. The project’s aim was to deliver a novel, ambitious perinatal platform to efficiently evaluate the clinical- and cost-effectiveness of multiple comparisons concurrently. Therefore, this article differs in structure from a conventional research article. Our intention is to describe our methods and sources in sufficient detail, so that others undertaking a similar project would benefit from and be able to repeat the research.
The collective vision of the Tiptop (Testing interventions in pregnancy and the newborn, trials in one platform) collaboration was to develop all aspects of project delivery for an ambitious and novel trial platform infrastructure, which would efficiently evaluate multiple interventions in perinatal medicine.
The ambition of this accelerator award was to create an efficient open platform with maximum stakeholder buy-in, and randomisations open to women and their infant(s) at different timepoints, allowing multiple interventions to be evaluated within a shared infrastructure (illustrated in Figure 1).
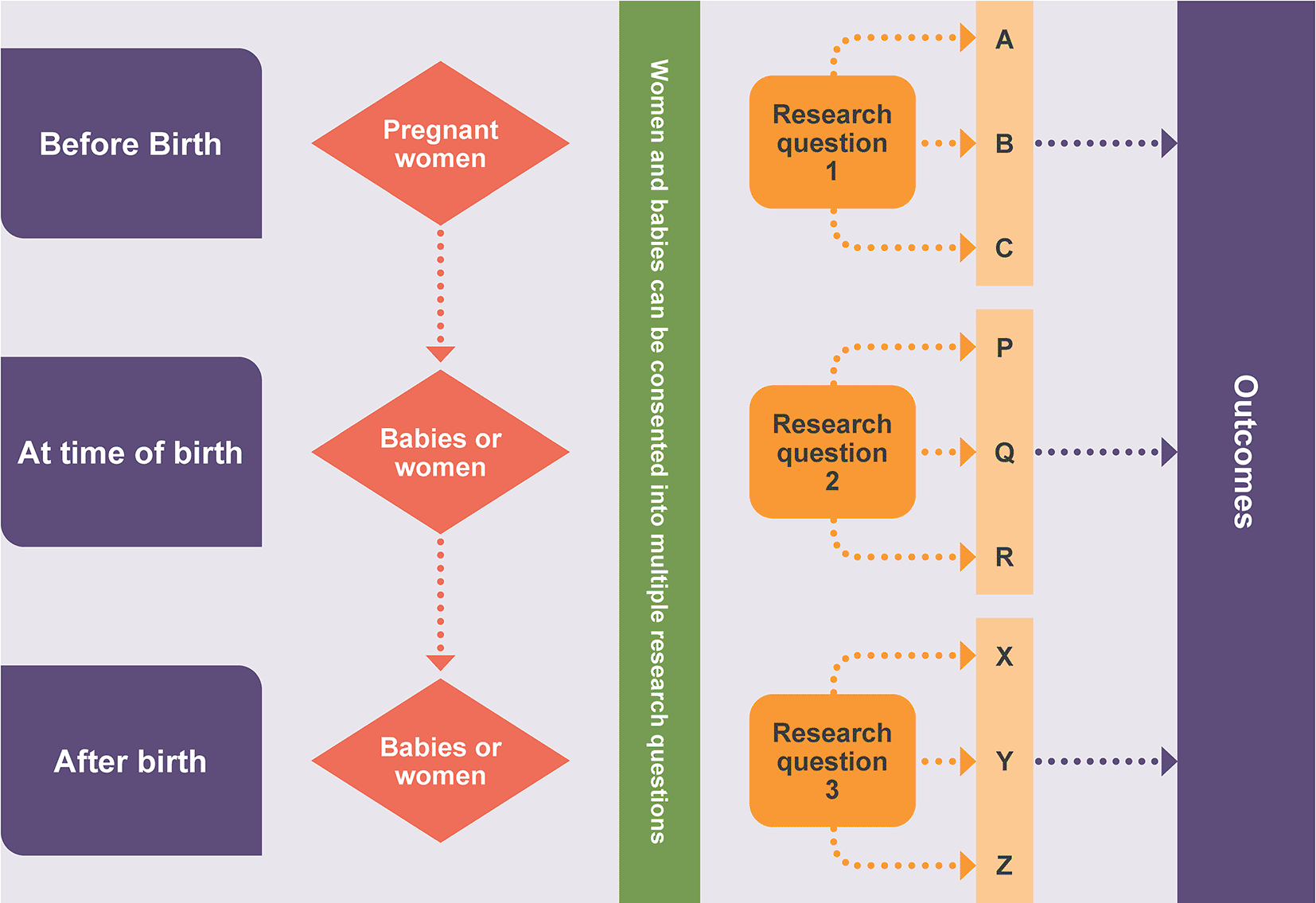
The platform would allow women and their babies to be recruited into more than one research question at any point in time along their care pathway. For example, a pregnant woman could be recruited into a trial investigating methods to diagnose diabetes in late pregnancy. Then at the time of birth, if the baby is born prematurely, the baby may be eligible for a trial investigating how to manage the umbilical cord. If that baby is then admitted to a neonatal unit, they may be eligible for a trial investigating the fortification of milk feeds. These trials would be conducted within a shared infrastructure, enabling efficiencies to deliver evidence to the bedside far more quickly.
The accelerator award enabled us to build sustainable partnerships between clinicians, methodologists and statisticians from across the UK with expertise in the delivery of perinatal and platform trials, and to focus on key operational, clinical, methodological, feasibility, parental/familial, and inclusivity aspects of the platform design.
Tiptop would enable:
• Evaluation of research questions relevant to pregnant women, and women and babies during and after birth;
• Multiple research questions to be addressed simultaneously within an efficient infrastructure;
• Enrolment (i.e., consent and randomisation) of women and babies into trials within the platform at any point along the care pathway during pregnancy, and during and after birth;
• Women and babies to be enrolled into more than one trial in the platform;
• The addition of new interventions to existing trials, as well as the addition of whole new comparisons addressing new research questions;
• Clinical investigators to lead on research comparisons (i.e., an ‘open’ platform).
Problems that occur during pregnancy, birth and the newborn period may result in death or survival with significant life changing morbidity for the mother and baby, with implications for the family and wider society.
A current UK Government policy focus is targeting reductions in deaths of mothers and babies and preventing brain injuries in babies.1–3 Reducing this burden is also important globally as neonatal disorders remain the leading cause of life years burdened by disability worldwide.4 Stillbirths still occur in ~6 in 1,000 UK births and ~3 in 1,000 UK newborn babies die in the first 28 days.5 Addressing disparities in UK maternal and perinatal outcomes is a further priority (e.g., worse maternal and perinatal outcomes for Black and Asian British women).5 Protecting a mother and partner from such psychologically traumatic events is also particularly important.
The two groups of babies most at risk of death or brain injury potentially resulting in lifelong impairment are those born following a traumatic birth and/or born very prematurely, before 32 weeks of gestation. Improving outcomes for these vulnerable groups is the focus of this platform, linking the pregnant woman-mother-baby dyad to enable a range of interventions to be evaluated in a highly efficient manner.
Combining these maternity and neonatal research questions in a platform would enable efficient information sharing, streamlined consenting for parents, multiple and transparent care options (identified by our extensive patient and public involvement work as particularly important to parents) and common data collection for both short- and long-term outcomes.
There are many unanswered perinatal research questions that this platform can address.6–8 The neonatal questions have been rated as important top 10 priorities by healthcare professionals and the public in a priority setting partnership.7 Recent data indicates that birth injuries associated with impacted fetal head in the UK are now as common as those associated with shoulder dystocia; our team was commissioned by NIHR to undertake a scoping study in this area (NIHR17/75/09).9–11
1.2.1 Why is a platform trial design required and appropriate?
Despite recent improvements in obstetric and neonatal care and increasing survival rates among extremely preterm births, there has, as yet been no tangible reduction in the proportion of infants surviving without neurodevelopmental disability.12 Indeed, some consecutive cohort studies show that educational outcomes and quality of life may even be declining over time. It is thus likely that improvements in survival and neurodevelopmental outcomes will vary by gestational age at birth, be incremental, requiring multiple, simultaneous interventions and approaches to perinatal management.13,14 Outcomes which are considered important to pregnant women/parents have been identified and could be easily, validly and reliably measured for platform outcomes.15 In addition, there are several valid ways for assessing infant development, so one of our tasks was deciding the best way to use these and measure them in a platform. Therefore, the perinatal field is fertile ground for platform trials.
Multiple clinical research questions exist that could be simultaneously addressed in an efficient trial structure and design. Importantly, a platform design would provide the opportunity to investigate multiple interventions at different time points across pregnancy (antenatal/peripartum/postpartum).
Using a platform design would result in a coordinated and far timelier evaluation of candidate interventions (pharmacological and non-pharmacological) and expedite decision-making to influence policy. More efficient evaluation of vulnerable participant populations (e.g., preterm babies), gives us the potential for the biggest impact most quickly. Rapid evaluation would also help eradicate the use of less effective, potentially costly, and invasive interventions. Combined, this would maximise impact on the participant population with life-long potential benefits to families, society and the NHS per se, not only by bringing beneficial interventions more quickly to the bedside, but also providing them with greater access to novel treatments. This was essential if we were to meet ambitious targets outlined by Department of Health and Social Care (DHSC).
Therefore, we proposed a novel and efficient perinatal platform (as opposed to multiple separate randomised controlled trials), co-developed with our PPI members with maximum stakeholder buy-in, on which multiple randomised comparisons can be performed. It was designed to improve care in a high-priority area highlighted by the UK Government and of global importance.
The platform would streamline recruitment of women and babies into multiple trials in a network of sites and use shared: IT infrastructure; consent procedures; outcome assessments and data capture; specialist expertise across Clinical Trials Units. It would allow the addition/removal of interventions (e.g., early stopping for futility or harm as trial adaptation across all research questions) and new comparisons, which would allow testing of the most promising interventions, as well as rapid changes to the standard of care. This approach would represent a step change in the efficient delivery of evidence. Future comparisons would be added, benefiting from the infrastructure – requiring less resources and faster setup and completion.
There are many unanswered perinatal research questions requiring confirmative trials. Consumers and scientific groups, including the Cochrane Collaboration and the International Liaison Committee on Resuscitation (ILCOR) highlight priorities relating to the optimal management of at-risk mother-baby dyads at the time of birth.6–8
Transparency and efficiency in the review and adoption process of new comparisons would be paramount to ensure buy-in from potential investigators who wish to lead a new comparison. An Independent Scientific Advisory Group (ISAG), including national and international experts, would play a crucial role in the selection of new comparisons. The ISAG would provide a transparent and equitable evaluation process for research questions seeking to be included in the platform in future, using prespecified criteria.
1.2.2 The importance of the connection between women and their babies
This platform had the potential to join up areas of medical research that have been, until now, unnecessarily conducted in isolation, despite the obvious connection between a pregnant woman and her infant(s). Neonatal and infant outcomes are critical to maternity and obstetric interventions, and neonatal care influences outcomes of maternal and obstetric interventions – they are intrinsically linked. Hence the need for a platform that considers maternity and neonatal care together.
In order to create, maintain and support such an ambitious endeavour, a large multidisciplinary team of stakeholders and experts from multiple institutions was required. The infrastructure would resemble a “virtual Clinical Trials Unit”, each bringing their individual specialisms, whilst being able to be flexible and provide them interchangeably. Such a team would need to be well organised, and communications would need to be clear, inclusive and frequent.
Several institutions contributed to the design, conduct and oversight of the platform, including four universities (with three UKCRC-registered Clinical Trials Units (CTUs) embedded) and five research active NHS Trusts ( Figure 2). The collaboration built upon existing networks of academic institutions which had successfully delivered numerous landmark, multicentre, NIHR-funded clinical trials in the perinatal field over the last 20 years.16–30 Experience gained from involvement in the RECOVERY platform trial (https://www.recoverytrial.net) and in setting up the PROTECT-CH trial (https://www.protect-trial.net) and experience in running adaptive trials, demonstrated the need for a large multidisciplinary team across multiple institutions. Key challenges identified include increased complexity, statistical and non-statistical methodological considerations, impact on coordination/organisation, and explicit pressures on capacity and training within CTUs.31 This is why we involved three CTUs (NCTU at the University of Nottingham, NPEU CTU at the University of Oxford, CTRU at the University of Sheffield). Each CTU led (with some overlap and flexibility) on key aspects; e.g., NCTU on trial delivery, methodology, Patient and Public Involvement and Engagement (PPIE) and data flows, CTRU on statistical methodology and NPEU on trial governance, IT systems and health economics.
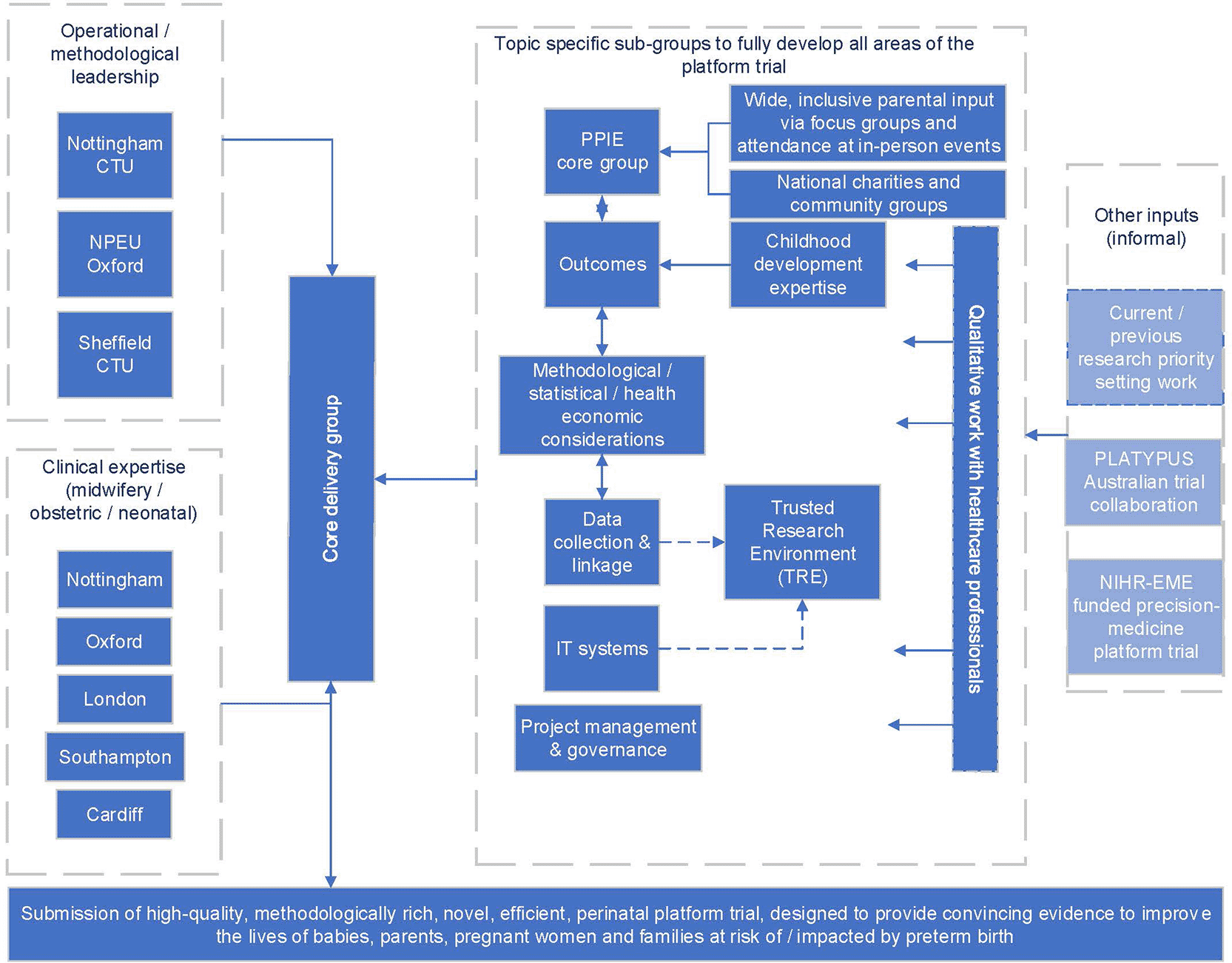
Figure 2 illustrates the final configuration of the Tiptop collaboration – it required a complex integration of multiple institutions and functions, arrived at following several iterations.
Our award was jointly led by senior researchers at two CTUs (EJ, NCTU & PH, NPEU CTU) and a senior clinician (JDo). They worked closely together throughout the grant to provide overall leadership and to ensure deliverables were met on time and within budget. They had weekly virtual “catch-up” meetings, convened by an experienced project coordinator (FM). Co-investigators EM and KS provided operational leadership and oversight at their respective CTUs, working closely with the project leads. Statistical expertise in adaptive trial design and conduct was provided by experienced methodologists at Sheffield CTRU. The experienced project coordinator (FM) worked closely with the methodological and operational leads to ensure the smooth running of the project.
A core group comprising individuals with methodological, statistical and operational (Nottingham, Oxford, Sheffield) expertise met monthly, alternating between virtual and in-person meetings (the Core Working Group). Meetings were agenda driven, with key actions and deliverables noted and communicated to avoid delays in project management. Clinical co-investigators (CG, JDo, KW, MS, SO, MK, CR, JS) led on all clinical aspects (meeting every 6–8 weeks initially, and then every 2–4 weeks when developing the grant submission) and with regular two-way communication with the Core Working Group. Subgroups, discussing specific clinical, methodological and statistical aspects of the platform were set up, comprising members of the co-investigator group; these met regularly and fed back to the Core Working Group. The core PPIE group met every 6 weeks to discuss progress and was responsible for the three proposed stages of parental contribution, i.e., (i) Listening and opinion seeking; (ii) Development of an inclusive PPIE strategy, and (iii) Communications/trial materials. The whole co-investigator group met four times in-person, for a full day, over the 12-month period for key-decision making and ensuring deliverables were met. On the first two occasions, the focus was on refining the clinical research questions and associated trial methodology, plus developing and finalising the perinatal platform clinical trial application to be submitted in November 2023.
The work required to deliver the platform involved:
• Overall leadership through platform delivery leads [JDo, EJ] as part of a multidisciplinary executive group [PH, KW, CR, JS, EM, KS, MD, MB, AK, RP, SJ, platform delivery manager];
• Clinicians (all with large, multicentre perinatal/obstetric trial chief investigator experience) representing the perinatal period (antenatal/ peripartum/postpartum), namely obstetrics, midwifery and neonatology [KW, NS, JDo, CR, SO, CG, JS]; public health [MK];
• Perinatal trials expertise – all clinicians plus EJ, PH, EM, CP, JDa, LF, AK, KS, OR-A, SJ, RP; Medical statistician with specific expertise in adaptive and non-adaptive trials [MD], and clinical trial methodologists with experience in perinatal trials [EJ, PH, CP, MB, EM];
• A health economics specialist in conducting economic evaluations of perinatal interventions from a family perspective [OR-A];
• A specialist in child development whose expertise includes evaluation of neurodevelopmental outcomes for preterm infants and on developing outcome assessments for use in clinical services and research [SJ];
• Platform trial delivery experience gained from several team members being involved in the design, randomisation/IT systems, conduct, monitoring, statistical analysis and oversight of the RECOVERY COVID-19 platform trial [EJ, AK, DM, RW, CG, CR, MK];
• Data capture (routine and bespoke), curation/(Trusted Research Environment) [JDa, LF, PQ];
• Patient and Public Involvement and Engagement (PPIE) from the outset [RP, EM]; Patient and Public Involvement was led by RP, an independent PPI consultant who led the public advisory group [MA, NL, CMcC, ES, HV, JW];
• PPIE was also provided by charities: Bliss (a UK-based charity for infants which supports the families of babies in neonatal care and works with health professionals to provide training and improve care for babies) [represented by CL-D] and NCT (National Childbirth Trust is the UK’s largest charity offering information and support in pregnancy, childbirth and early parenthood) [represented by HJ];
• Qualitative/process evaluation expertise [PL];
• Capacity building and platform sustainability was maximised by adopting comparison co-chief investigators [CG, EM, SO] and inviting early career researchers [KC, HF, CF, KH, JO’S, MR, AS] as comparison co-investigators.
The creation of the platform depended upon successful completion of several interlinked but distinct aims and objectives – several working groups were quickly established to work on the tasks required to deliver the project (detailed in Table 1). Those working groups took responsibility for the project deliverables (see Figure 3).
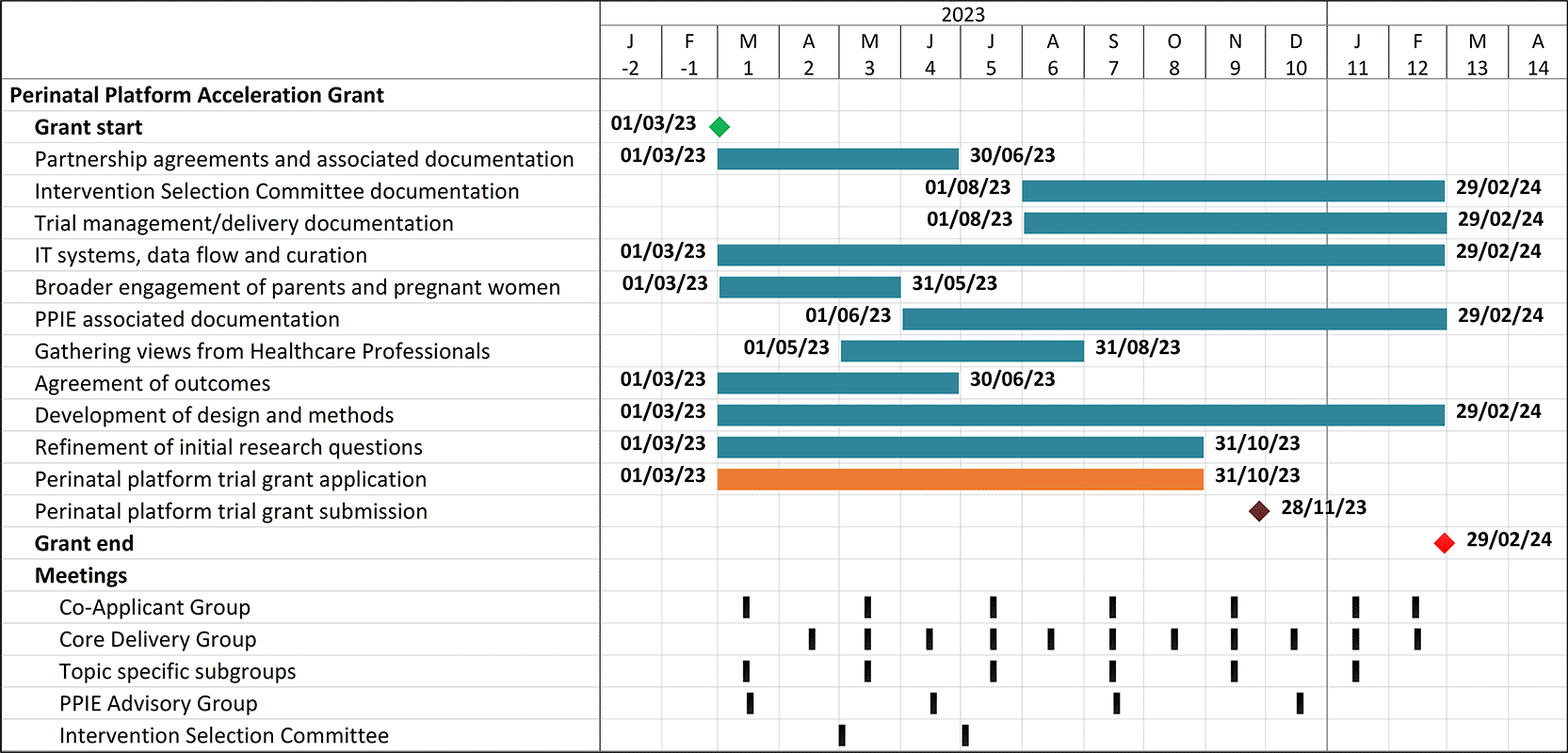
Essential elements in developing the platform infrastructure and trial design plus meeting the key objective of submitting a high-quality grant application to the November 2023 commissioned call included:
• Developing the research questions;
• Platform governance and partnership agreements/oversight arrangements (e.g., holding sponsor meetings);
• Engagement with parents and pregnant women on trial design, procedures (e.g., consent/enrolment, follow-up schedule) and interventions;
• Consulting with the perinatal clinical community to ensure clinician buy-in;
• Corresponding design and methodological work (including extensive statistical simulation), agreement of outcomes (including PPIE), and economic evaluation;
• Developing IT systems and framework.
These aspects are covered in more detail in this section. All of these elements required iterating towards the ‘end product’ and sometimes this was not predictable, in which case we needed to be flexible and agile both in our discussions and actions. Likewise, several of these elements were conducted in parallel.
One of the key deliverables of the accelerator award was to submit a high-quality grant application to the November 2023 commissioned call.
To achieve this, we needed to refine clinical research questions, building upon prioritised, consensus stakeholder views and opinions7 to decide and shape our research plans. Specifically, each research question required a clear definition of the:
• Population: which individuals the question needs answering for;
• Intervention(s): which one (s) should be evaluated;
• Comparator: what standard of care these interventions would be compared against, and;
• Outcomes: the metrics by which these would be judged.
The Population, Intervention, Comparator and Outcome are referred to as the ‘PICO’, and are a central part of framing research questions in a clinical trial.
The co-investigator group, which included prominent clinicians/clinical trialists with expertise covering obstetrics, midwifery, neonatology and child development alongside parents, charities, methodologists and trial delivery specialists, was central to this process. A PICO subgroup was formed to discuss potential research questions – it continued to meet regularly from the start of the accelerator award in order to develop the first perinatal platform trial proposal. As part of the accelerator award application, the co-investigator group had already identified a number of potential interventions covering the perinatal period.
As part of the discussions related to the accelerator award application, the PICO group had identified three possibilities at different time points across pregnancy, including:
• Antenatal: timing/dose/repeat courses of antenatal corticosteroids for women with threatened preterm labour (to allow fetal lung maturation and for prevention of neonatal brain injury);
• Peripartum: multiple approaches to cord management at birth in infants <28 weeks’ gestation – options to compare included resuscitating with the umbilical cord intact, quickly clamping and resuscitating after cutting the cord, or milking the blood from the cord before or during resuscitation (to increase preterm survival and prevent neonatal brain injury);
• Postpartum: different approaches to respiratory management (e.g., location/timing, doses and frequency of caffeine administration, maternal caffeine) and nutrition in the preterm infant (e.g., early fortifier vs late fortifier).
To refine the clinical questions, we conducted some feasibility/methods work alongside consulting our parent advisory group and wider PPI and healthcare professional communities. It became apparent, that while clinically important, a trial of antenatal corticosteroids would be impractical in terms of sample size, time taken to recruit such a large sample, and cost. Therefore, this option was demoted.
However, given that our unique selling point was the connection between mother and baby, we rapidly assessed another research question addressing the prevention of maternal morbidity, improvement of infant survival and prevention of hypoxia and infant brain injury, which was the evaluation of interventions to manage an impacted fetal head. The sample size estimates were promising in terms of feasibility, and the research question had undergone extensive, funded feasibility work. Therefore, this was considered ‘HTA-ready’, and was adopted as a research question for inclusion in the Stage 1 application. The initial research objective was to evaluate two techniques for managing an impacted fetal head at caesarean birth when labour has resulted in the head becoming impacted in the pelvis: the fetal pillow, and the standard approach, the vaginal push technique. Other candidate management techniques could be added in future, with sufficient evidence of a ‘signal’.
Discussions with the PICO group regarding the multiple approaches to cord management established that two of the interventions were ‘HTA-ready’. Therefore, in babies born before 32 weeks’ gestation who need resuscitation, evaluating deferring clamping the umbilical cord for between 3 and 10 minutes compared to early clamping in the first minute from birth, was adopted as a research question, following assessment of feasibility. This topic had been identified as a priority in a national priority setting exercise.7
Subsequent discussions regarding greater potential efficiencies for the platform identified another top priority for research into preterm babies regarding respiratory support.7 The participant population would dovetail very nicely alongside the cord management research question and so, following assessment of feasibility, a third research question was adopted by the PICO group. Namely, in babies born before 32 weeks’ gestation who do not need resuscitating (i.e., ineligible for early clamping), evaluating the use of early nasal Continuous Positive Airway Pressure (nCPAP) compared to Non-Invasive Positive Pressure Ventilation (NIPPV).
Lastly, the agile and adaptive nature (by design) of the platform meant that the existing research questions might have evolved over the course of the trial. For example, new interventions could arise that might be added to an existing comparison, or new comparisons altogether may be proposed and added, under the right circumstances. We describe the process for this in the subsequent section.
Our aim was to develop governance processes for selection of the initial and future interventions by collaborative, multistakeholder oversight of a perinatal platform. Such an endeavour also required novel, collaborative thinking with respect to sponsorship considerations. Solutions to these issues are explored here.
3.2.1 Developing the initial research questions
The limited time between the award’s start date and the HTA deadline for the Stage 1 application of the proposed platform meant that the first research questions and their PICOs needed to be developed within the co-investigator group. Whilst this was inevitably more restrictive than the wider collaboration described above, the group took several steps to ensure that prioritisations were made as objectively as possible. A PICO subgroup was created, chaired by a member of a CTU whose institute was not involved in developing the research questions. The group developed a formal terms of reference document and criteria for selection (see Table 2). The proposal was ambitious, comprising a series of interlinked trials unified within a national collaboration which could be adapted to make the best use of human and capital resources.
3.2.2 Developing subsequent research questions
The platform would allow the addition of interventions to those already being evaluated, and discontinuation of interventions demonstrated to be beneficial, futile (‘fruitless’) or harmful. Whilst the latter can be predefined by stopping decision rules, the process of prioritising new research questions and comparisons requires careful oversight.
Once the platform had been set up and launched with the initial set of research questions, the intention was to add studies onto the platform based on input from the wider research community, ensuring an open platform within which investigators (outside of the original co-investigator group) could lead research questions. Members of the platform delivery team would maintain a presence to ensure appropriate proposals were submitted, including at a minimum, an ongoing internet presence and conference presentations.
A number of prioritisation setting exercises relevant to perinatal research are underway or have been published that would be key to ensuring sustainability of the platform, and that priority areas would be considered. One key piece of work is a national consensus among healthcare professionals, researchers and parents in the UK, led by a platform collaborator and national organisations (Neonatal Society and British Association of Perinatal Medicine) to prioritise research questions suitable for perinatal platform trials.7
Transparency, efficiency and fairness in the review and adoption process of new research questions would be paramount to ensure buy-in from potential investigators across the UK, specifically those outside the institutions involved in the proposal, who would wish to introduce and lead a comparison within the platform. With this in mind, and based on similar existing frameworks, we developed an oversight and decision-making framework by which interventions and trial adaptations could be selected.32 The framework would involve input from two groups:
1. Independent Scientific Advisory Group (ISAG): a committee of experts representing key clinical areas in perinatology, independent of the key collaborators on the platform to ensure fairness of review;
2. Tiptop Executive Committee (TEC): the platform collaborators involved in the key functions of the platform, including platform leads, methodologists, statisticians, PPI and trial delivery, with the in-depth knowledge of the platform required to assess the applicability of the proposed research question to the platform methodology, infrastructure and to PPI.
To ensure an efficient and timely review process, frequent meetings of the ISAG to review proposals would have been required, which was considered to be overly burdensome. It was therefore decided that all proposals would be reviewed by members of the ISAG electronically, and a timely response would be expected, with central monitoring and adjustment of workload. An inaugural meeting followed by annual meetings of the ISAG were planned to agree and review membership and processes. The process would allow for conflicts of interest if, at any time, a member wished to submit or co-submit their own research question to the platform.
To ensure transparency, Terms of Reference and the review process would have been made available on the Tiptop website, and written feedback on submitted proposals provided by the TEC.
An ISAG was established, comprising national and international representation of key clinical expertise and all with experience in RCTs (3 neonatologists, 2 obstetricians, 2 midwives, and 2 neonatal nurses).
Selection of new research comparisons/questions would follow a two-stage process:
Stage 1: The ISAG would assess potential research questions proposed by investigators (using a proforma) against prespecified criteria relating to scientific merit and acceptability (see Table 2). The ISAG would provide recommendations to the TEC on whether a research question should be considered for adoption onto the platform. The ISAG would be sent proposals electronically, with written feedback and recommendations provided from each member for consideration by the TEC.
Stage 2: The TEC would meet regularly to review recommendations for adoption against additional prespecified criteria relating to incorporation within the existing platform (see Table 2) and provide written feedback to potential investigators.
3.2.3 Sponsorship
Understanding and developing a novel and efficient governance and sponsorship structure was integral to this accelerator award, due to the large numbers of institutions involved. We wanted to ensure the sponsorship structure met the governance requirements for adhering to clinical trial regulations, alongside balancing institutional requirements for Chief Investigators and ensuring equity across institutions. The governance ‘subgroup’ met with several stakeholders across various institutions to develop a proposed sustainable long-term framework for sponsorship, which would have helped ensure rapid setup of research questions.
We gained insights from trial teams and sponsors running other successful platform trials and attended structured platform training sessions. We spoke with a range of stakeholders such as Chief Investigators, Trial Managers, Directors of Research Governance and colleagues from Joint Research Offices to ensure considerations were included from multiple disciplinary teams.
The group concluded that the Tiptop platform would have a pool of two sponsors based within the Clinical Trial Units directly involved in trial management of the research questions (University of Oxford and University of Nottingham). The initial research questions and any subsequent intervention arms and research questions would be adopted by one of the sponsor teams. A joint sponsorship model was not deemed suitable due to the complex governance and indemnity arrangements.
Efficiencies would be retained by agreeing core Standard Operating Procedures (SOPs) and shared template documentation such as protocol, Patient Information Sheet and Consent Forms. A dual sponsorship review would have taken place for the first three research questions. New research questions could be deployed quickly by using the agreed template documentation suite and adopting the Tiptop ‘brand’.
The pool of sponsors would have been joint data controllers, to allow for an agile sponsorship model and data sharing efficiencies for shared IT and Trial Management teams.
To ensure an open access platform for all potential future investigators, a Chief Investigator contractual agreement, developed by the University of Oxford would be used for any Chief Investigators not holding substantive employment within the sponsorship pool.
The insurance teams and underwriters were made aware of the Tiptop platform, and insurance costs were included to ensure that all sponsorship responsibility would be covered using this novel sponsorship model.
The Research Governance, Ethics & Assurance Team and Joint Research Offices were key to developing this complex agile sponsorship framework and were keen to implement lessons learned from platform studies that were set up in response to the COVID-19 pandemic.
Our aim was to work with parents, pregnant women and national charities to ensure integral public involvement in the development of the platform, including creation of a robust, inclusive PPIE strategy for the platform. Essentially, putting parents and their infants at the heart of everything we did was of crucial importance.
For such a large platform trial, with the future aim of involving thousands of women and their babies, and health professionals around the UK, it was important to consider the views of this broad community. We had a two-pronged approach to community engagement, involving patients and members of the public and healthcare professionals which we now describe in turn.
3.3.1 Patient and public involvement – engagement with parents and pregnant women
First, we established a diverse patient and public involvement group from the outset. This was led by an independent parent and public involvement consultant (RP) and a member of the team with substantial experience in PPI activities (EM). To recruit women to join the group, we drew upon existing national contacts and placed simple adverts on social media channels. Our PPI group is diverse in ethnicity, location of the UK where they are based and their birth experiences and outcomes. Since initially the research questions focused on questions relevant to prematurity, all members had experience of preterm birth. Latterly, with the introduction of a third research question, not relating to prematurity, the group was extended to include a woman with lived experience of an impacted fetal head.
PPI members received ‘terms of reference’ describing the role, and expectations around the role, to which they agreed. The group also discussed the process for which they could seek support, should any discussions become distressing or upsetting, given the sensitive nature of the clinical area. PPI members were advised to ‘step away’ from discussions should they find them too difficult and reach out to other members of the group for support or directly to RP/EM if they wished to, by sending a private message. They were also signposted to other groups that provide support to parents. The group met on Zoom at regular intervals over the 12 months and, with their permission, a WhatsApp group was set up at the beginning for fast and easy communications. A Dropbox account was also utilised to share documents, as this was the virtual platform that most PPI members felt most comfortable with and had access to. The first Zoom session was a training session to ‘set-the-scene’, explain more about PPI, the aims and objectives of the study and agree how the group would work together. The PPI group worked closely with the research team throughout the award, contributing to key decisions, and were invited to relevant team meetings.
Whilst we had established a diverse PPI group, we recognised the need for wider parental engagement, and, with the PPI group, agreed that parent-led meetings would be held with other parents to gauge broader views. Once initial research questions had been agreed, eight parent-led meetings were organised and held on Zoom, during 2023. To recruit additional parents for parent-led meetings, we posted adverts on social media platforms and utilised connections via our charity partners, Bliss and NCT.
Each meeting was facilitated by a member of the PPI group, with a second member monitoring ‘chat box’ activity and feeding points into the discussions. RP also attended in a support role. The PPI group met before the first wider parental meeting was held to discuss content and agree on how they would manage the sessions. Daytime and evening sessions were available, recognising that individuals have different availability and commitments. Four topic areas were covered (one evening and one daytime meeting per topic): research questions, approaching/screening parents, consent, and outcomes. Sessions were recorded and written notes shared with the PPI group, via the Dropbox account. Sixty-one people joined at least one parent-led meeting. RP maintained a register of attendance and attendees were paid for their time, in line with NIHR guidelines for PPI activities. Key themes that arose from the discussions were the need for early engagement and communication with parents and the importance of having an inclusive approach to recruitment.
In addition, the PPI group met specifically to discuss a communication strategy throughout the platform trial and the approach the group should take to inclusivity, specifically working through the NIHR INCLUDE ethnicity worksheets, with a view to considering wider aspects of inclusivity than just ethnicity.
3.3.2 Consulting with the perinatal clinical community to ensure clinician buy-in
Our aim was to qualitatively explore the views of a range of healthcare professionals (HCPs) about the design and conduct of a UK-wide perinatal platform trial.
Five online discussion groups, led by EM and PL, were organised for July and September 2023. The meetings were organised over lunchtime or at 08:30 to facilitate attendance by clinicians, avoiding morning or afternoon clinic or theatre sessions. To publicise the discussion groups, adverts were placed on X (formerly Twitter) by members of the research team and CTU X accounts. In addition, clinical members of the research team and CTU leads sent emails to their extensive network of clinical contacts, and information was also shared by the British Association of Perinatal Medicine (BAPM), the Royal College of Obstetricians and Gynaecologists (RCOG), the British Maternal Fetal Medicine Society (BMFMS), the UK Preterm Clinical Network and the British Intrapartum Care Society (BICS). Our aim was to reach a diverse group of healthcare professionals, of different levels of experience and seniority, across the UK, including obstetricians, neonatologists, midwives and neonatal nurses. Healthcare professionals who saw the advert and were interested in attending were asked to email a central mailbox to register their interest in attending and were then sent a calendar invite with link to a Microsoft Teams meeting, to one of five sessions of their choice.
Each session was facilitated by EM, PL and at least one clinical member of the research team. A standard slide set was developed, which included background information about what platform trials are and why a perinatal platform trial is important, followed by slides outlining the team’s thoughts on the initial research questions to include in the platform. These were presented using a PICO (population, intervention, comparator, outcome) format. Attendees were encouraged to be interactive and ask questions from the outset. After each research question was presented, questions and discussion was invited. For each of the research questions, attendees were asked whether they felt the research question was relevant and whether they would be happy to be randomised as a participant.
A total of 56 health professionals attended an online discussion group. This included 13 obstetricians, 17 neonatologists, 14 midwives, 8 neonatal nurses and 4 other healthcare professionals (a preterm birth prevention lead, a pharmacist, a NIHR doctoral research fellow, and a maternity/neonatal research programme manager). These healthcare professionals were from England, Scotland and Wales.
3.4.1 Choice of outcomes
We defined a set of core outcomes for each proposed research question within the platform. Common outcome measures were defined across comparisons, drawing on existing literature and published core outcomes. Ensuring that there was a cohesive underlying narrative for the whole platform was paramount, including the assessment of maternal, neonatal and infant neurodevelopmental outcomes.15,33 The outcomes of primary importance for one research question could, on occasion, conflict with the most relevant considerations to others. Where necessary, the impact of making trial adaptations would be based on more than one outcome.
3.4.2 Identification of common information and outcomes
To optimise efficiency of the proposed platform, we identified outcomes common to the three different studies with the aim to reduce duplication and redundant collected information, both for the bespoke and routinely collected data for the same population.
Table 3 shows the datasets needed and the corresponding data providers for each data source. Tables 4 and 5 detail the final information and outcomes needed (following an iterative selection process), the overlap between the three separate trials and the data sources used to obtain the information. Maternal short-term and long-term outcomes ( Table 4) were selected only for the Impacted Fetal Head (IFH) research question. Maternal long-term outcomes, including Post-traumatic stress disorder (PTSD)/maternal trauma, depression, anxiety disorders, severe mental illness, cervical insufficiency, tokophobia and sexual activity, were not available from routine data sources and therefore were to be collected through questionaries only. Infant outcomes ( Table 5) instead were partially overlapping between the three studies and all the information not currently available or not accurately recorded in routine data (RD) were planned to be manually collected (MC). However, in some instances, we also planned to combine manually collected data and routine data to augment the details of the needed information. Economic outcomes ( Table 6) were mainly common to the three studies.
3.4.3. The importance of long-term neurodevelopmental follow-up
Relative to children born at term, children born very preterm are at increased risk of a range of developmental problems and disorders which can have adverse impacts on health and wellbeing across the lifespan. Although a small but significantly increased proportion of children have neurosensory disabilities, the most common adverse outcomes are cognitive impairments, present in up to 50% of children born extremely preterm.12 In the long term, these can include difficulties with attention, working memory, visuospatial skills and executive functions which can have a major impact on attainment at school, limiting life chances.34,35 To date, consecutive cohort studies show no significant reduction in the prevalence of neurodevelopmental impairment among children born extremely preterm despite increased survival and improved neonatal care.12,14 Thus, improving neurodevelopmental outcome remains a key priority for neonatal medicine. Moreover, long-term developmental outcomes are considered of high importance to parents and are key components of core neonatal outcomes.15
Developmental follow-up in early childhood is crucial to identify children at risk of long-term impairment to enable early intervention and to provide routine data for clinical audit. Routine clinical follow-up is typically carried out at 24 months of age for very preterm born infants when a standardised developmental assessment is recommended to be used to assess cognitive and language development, particularly as routine clinical assessment has been shown to lack adequate sensitivity in detecting impairment in cognition and communication domains.36,37 However, although 71% of children born preterm and admitted for neonatal care in 2018 had a routine 2-year clinical assessment, in only half of these was a standardised developmental test performed.38 Thus, routine data was not considered sufficient for the complete and accurate assessment of 2-year neurodevelopmental outcome in the context of the platform.
For both neonatal questions, neurodevelopmental outcomes were considered of key importance as both interventions may have impact on long-term developmental outcome, either via a direct influence on neurodevelopment or via reduction of neonatal morbidity such as intraventricular haemorrhage (IVH - bleeding in the brain), or bronchopulmonary dysplasia (BPD - long-term breathing problems in premature babies). As such, long-term follow-up was proposed to be carried out when infants reach 24 months of age corrected for prematurity, for which the key secondary outcome chosen was survival without moderate to severe neurodevelopmental impairment. For the obstetric research question, there are no existing data on the effect of IFH on children’s developmental outcomes despite the risk for neonatal morbidity, potential for adverse impact on brain development and the potential differential impact of the proposed interventions, thus this was considered clinically important to assess.15 Therefore, for all three trials, the same validated, parent-completed questionnaire with robust psychometric properties was proposed to be used to assess neurodevelopmental impairment among survivors. Neurodevelopmental impairment was defined as the presence of any one or more of cognitive impairment (Parent Report of Children’s Abilities-Revised (PARCA-R) non-verbal cognitive scale standard score < 70); language impairment (PARCA-R language scale standard score < 70); gross motor impairment (unable to walk and/or sit independently); hearing impairment (hearing loss corrected with aids, or some hearing loss not corrected by aids, or deaf ); visual impairment (reduced vision uncorrected with aids, or blind in one eye, or blind/can perceive light only). These criteria are commensurate with recommendations for the classification of health status at 2 years and include core neonatal outcomes of importance to professionals and families. Survival information would be obtained from routine data. As neurodevelopmental impairment is significantly associated with the primary outcome in each trial (i.e., adverse neonatal outcomes (impacted fetal head ), neonatal brain injury (cord management); BPD (respiratory management), use of the same outcome measure and the potential for data sharing across the platform provided greater efficiency and less burden for parents than the assessment of outcomes in multiple separate trials. The use of a validated parent-reported outcome measure also allows for cost-efficient assessment which has been shown to be acceptable to parents and used successfully in previous large-scale trials with good follow-up rates. Parental consent would be sought to allow the platform team to contact them to invite them their child to take part in future follow-up studies (subject to further funding) and to link their trial data with routine data on health and educational outcomes.
3.4.4 Health economics
The unique health economic opportunities that this platform trial presented were evaluated by the methodologists and led by our health economist (ORA) at the start of the project. Two key areas were identified that would need attention when developing health economics capacity within this platform: data collection processes and methodological considerations.
The implications of trial adaptations to ongoing trials and the addition of new comparisons to the platform in terms of data collection and the identification of relevant outcome measures to conduct economic evaluations were acknowledged. Data to undertake an economic analysis require information about healthcare utilisation and consequences to capture health impacts of treatment effect on health-related quality of life on babies, parents and other carers. Linking bespoke administrative databases (see Section 1.3.5.3) to the platform would facilitate the collection of healthcare utilisation for community and secondary care on a routine basis. Inevitably, for comparisons that require multicomponent interventions (e.g., umbilical cord clamping), a micro-costing approach would need to be followed. Patient-reported outcomes measures are not routinely collected in neonatal and maternal trials and would need to be captured using tailored patient questionnaires. In this platform, maternal and infant health-related quality of life would be collected using the EQ-5D-5L and EQ-TIPS instruments with any additional household information (e.g., employment-related data) also collected using participant questionnaires.
Methodological considerations of the platform included first the selection of the type of economic evaluation to conduct. Given the well-known limitations to capture data beyond 24 months in most of the neonatal and maternal trials conducted by the team behind this platform, it was agreed that all studies would present a cost-consequence analysis up to this time period as starting point. For neonatal trials, a cost-utility analysis using quality-adjusted life years (QALYs) as the main outcome measure with a lifetime horizon would also be presented. Being able to extrapolate beyond 24 months was identified as an additional methodological challenge and the health economics team proposed the development of a decision-analytical calculator that allows the extrapolation of the impact of treatment effect on costs and outcomes. Finally, the health economics team suggested evaluating the merits of value of information analysis to understand its role for specific trial adaptations (e.g., stopping early) and the assessment of decision uncertainty.
3.4.5 Statistical methodology
Our aim was to develop the most appropriate methodology for the platform. Trials conducted in antenatal/peripartum/postpartum settings raise several challenges, some of which are unique to the clinical area and necessitate a bespoke approach when designing a research platform. All trial designs have benefits and drawbacks, and we needed to carefully consider the implications of each different approach.
For each research question embedded within the platform, the trial design was informed by a combination of the views and opinions of multiple stakeholders including from patient and public involvement and engagement. For example, the unit of randomisation was informed by PPIE work which emphasised that, for open-label studies, parents felt strongly that siblings were randomised to receive the same treatment. The statistical performances of competing trial designs were explored via extensive statistical simulations which helped the team to decide on appropriate trial design aspects and to understand associated risks in decision-making.
We discussed how to leverage interim analyses to improve efficiency and feasibility, focusing on the outcomes to inform trial adaptations and when they would be assessed. Several different trial adaptations were considered depending on uncertainties and goals the research team hoped to achieve. This included whether changes would be made based on interim analyses to (a) stop the trial early (e.g., for futility/harm or efficacy) as soon as sufficient evidence is gathered to reach robust conclusions, (b) refine the target trial population to focus recruitment on those participants where the intervention shows most promise (adaptive population enrichment), and (c) update the number of mothers or infants required to answer the research questions (sample size re-estimation). We briefly considered, but then discarded, the possibility of response-adaptive randomisation designs in which participants are allocated in a way that favours interventions that show more promise, as it was impractical to implement in this context.
After consulting the clinical co-investigators and other stakeholders, it was agreed that, of these trial adaptations, early stopping for futility/harm would be likely beneficial and of interest both in the context of trial or treatment arm (s) early stopping. For example, for this platform evaluating multiple research questions, early stopping for futility/harm could be used to discontinue interventions (or research questions with interventions) showing a lack of promise or harm and prioritise resources for more promising interventions while safeguarding participants’ welfare.
Other trial adaptations were initially discussed but were not taken forward for different reasons. Table 7 provides a summary of the various trial adaptations that were considered as part of the platform development. While adaptive population enrichment was deemed to be valuable for the cord clamping research question, it was considered infeasible to further limit recruitment to subgroups of the trial population. There was a wealth of robust data to inform the initial sample size, and so uncertainty was not sufficient to necessitate sample size re-estimation for most research questions. While early stopping for efficacy could be useful, the interventions considered were believed unlikely to achieve the high bar of overwhelming evidence required to trigger early stopping. As such, benefits were viewed as very minimal at the expense of increasing the maximum sample size, undermining recruitment feasibility. Early stopping of intervention arm(s) for futility/harm in the context of multiple interventions (treatment selection) was considered for the cord clamping research question before viable study interventions were whittled down to one. However, we had planned to consider this as the platform evolved, where appropriate. Finally, for the IFH research question, the study team considered that there is a good chance for other competing interventions to mature for evaluation in the future. If this happens, these would be added on to the platform when recommended by the Independent Scientific Advisory Group and these would be considered ‘subtrials’, which would be designed focusing on pairwise comparisons and use of concurrent controls. Similarly, for each future research question added to the platform, we planned to revisit these different designs to assess their utility and feasibility. The IT and randomisation systems were designed appropriately with this in mind.
Each research question had its own statistical considerations. For example, clustering of outcomes around the mother was relevant for the respiratory research question as informed by the literature and discussions. The IFH research question had co-primary binary outcomes relating to the mother and infant that were viewed as important. The criteria for claiming evidence at the end of the trial and the nature of interim decision rules while dealing with these co-primary outcomes were informed by discussions with the clinical team. The intervention would need to show efficacy on both co-primary outcomes to claim superiority at the end and futility on both to trigger early stopping.11 Prior data indicated a very small correlation of 0.12 (95% confidence interval, 0.003 to 0.20) between co-primary outcomes, so we had planned to use a conservative approach without imposing this correlation.
3.4.6 Simulations demonstrating operating characteristics
Choosing the appropriate timing and frequency of interim analyses, as well as decision rules to trigger trial adaptations, is essential to the success of an adaptive trial to influence practice. Once we had identified trial adaptations of interest and some design parameters for each research question, we then carried out comprehensive statistical simulations to compare the performance of competing trial designs. Non-binding futility stopping rules were considered for flexibility in the interim decision-making process, allowing for consideration of all the available evidence. All simulations for the cord clamping and respiratory research questions were performed in R using ‘rpact’ v3.4.0 package.39 A bespoke program in Stata v18 was used for the IFH research question, as existing packages were not sufficiently flexible to permit correlated co-primary outcomes. We compared the frequency and timing of interim analyses and the decision rules applied at each interim analysis, while making different assumptions about the effectiveness of each intervention. This allowed us to assess the impact of these decisions on the operating characteristics of the design; for example, decision errors, the probability of early stopping, and the maximum and expected sample sizes. Simulation replicates for each scenario were chosen to achieve a reasonable Monte Carlo error within a feasible computational time.
Simulation work involved back-and-forth discussions with the clinical team and co-investigators, as well as presenting simulation results in lay terms to ensure all team members were able to contribute to making informed decisions. Simulation results were used to inform the choice of robust futility early stopping rules. As noted above, the clinical team preferred designs that focused on stopping for futility, but minimising the risk of (wrongly) stopping for treatments with small/moderate but important effects. They reflected that two of the interventions were unlikely to be harmful, and were relatively cheap and easy to implement in practice. In these cases, statistical simulations were therefore used to choose a design which minimised the probability of stopping early for lack of benefit when one existed. The simulation reports for each research question that were shared with the clinical team and co-investigators, that informed final decisions about appropriate adaptive designs, are accessible (via https://doi.org/10.15131/shef.data.25393327). Related statistical simulation codes are also accessible via GitHub (https://github.com/munyadimairo/TipTop-platform ).
3.4.7 Future research questions
The addition of future research questions would be guided by the potential impact on existing evaluations. Interventions aimed at pregnant women antenatally or during birth may affect (benefit or harm) the fetus, which in turn may affect subsequent intervention(s) aimed at the infant(s) or woman following birth. Whilst multiple interventions make it difficult to separate the effects of each individual intervention, we take the view that multiple interventions are common; and rather than assume these are small or “balance out”, we use a unified platform to incorporate such factors. We planned to use statistical methods that effectively re-weight the trial population, thereby allowing us to estimate each treatment’s effect in a hypothetical scenario where other interventions become more used, less used, or not used at all. In all cases, the estimand would be clarified using a defined target population, with statistical methods such as inverse probability weighted estimators to give insight into both population-averaged and conditional effects of interventions. Where new arms are added at the same time, this may be better considered as a nested trial with its own design with a concurrent shared control rather than an addition to an existing trial.40
3.4.8 Analysis
We planned multiple statistical analyses focusing on each of the primary comparisons of interest. Where necessary we planned to account for the natural clustering due to multiple births, depending on the estimand of interest. Where appropriate, the planned analysis would also make allowance for the fact that mothers and infants could be enrolled in more than one trial within the platform.
Interim analyses were to be conducted by a separate team of statisticians (based at a separate CTU) from the trial statisticians who provided ongoing input into the design and conduct of trials within the platform.
Our aim was to develop the IT systems and framework that the data collection methods, processes and data flows would operate within, in order to establish the platform.
To develop a flexible randomisation system that allowed randomisation of pregnant women and their infant(s), we planned to build upon the learning and IT systems of the RECOVERY trial (https://www.recoverytrial.net/). RECOVERY included an integrated system for randomisation, stock control for pharmacological interventions and data collection. We also intended to draw upon our experience from the data-enabled WHEAT pilot trial, where a randomisation system and data collection were embedded within an electronic patient record.41
Never before have women and their infant(s) been randomised into the same platform trial at different times, depending upon eligibility. Therefore, modifications to this bespoke system would have been required allowing (i) randomisation at the level of woman or infant(s), (ii) interventions to be added and dropped, (iii) randomisations to be performed at different stages across the perinatal period, and (iv) changes to the allocation ratio. The initial IT system development was to be prioritised based on the requirements of the first tranche of research questions, but agile modular programming would have allowed further modifications to incorporate important methodological developments.
To minimise the data collection burden for trial sites and families, we intended to maximise use of routinely collected data sources (e.g., the data warehouse for routinely collected neonatal data – National Neonatal Research Database) and minimise bespoke data collection. Data flow processes for collecting routine data across the UK would have been established and governance, curation, archiving and data sharing functionality set up within the Trusted Research Environment at the University of Nottingham.
3.5.1 IT Infrastructure
The IT infrastructure would be based on the NPEU CTU in-house applications (enrolment and trial administration) and the OpenClinica Clinical Database Management System (CDMS) (see Figure 4).
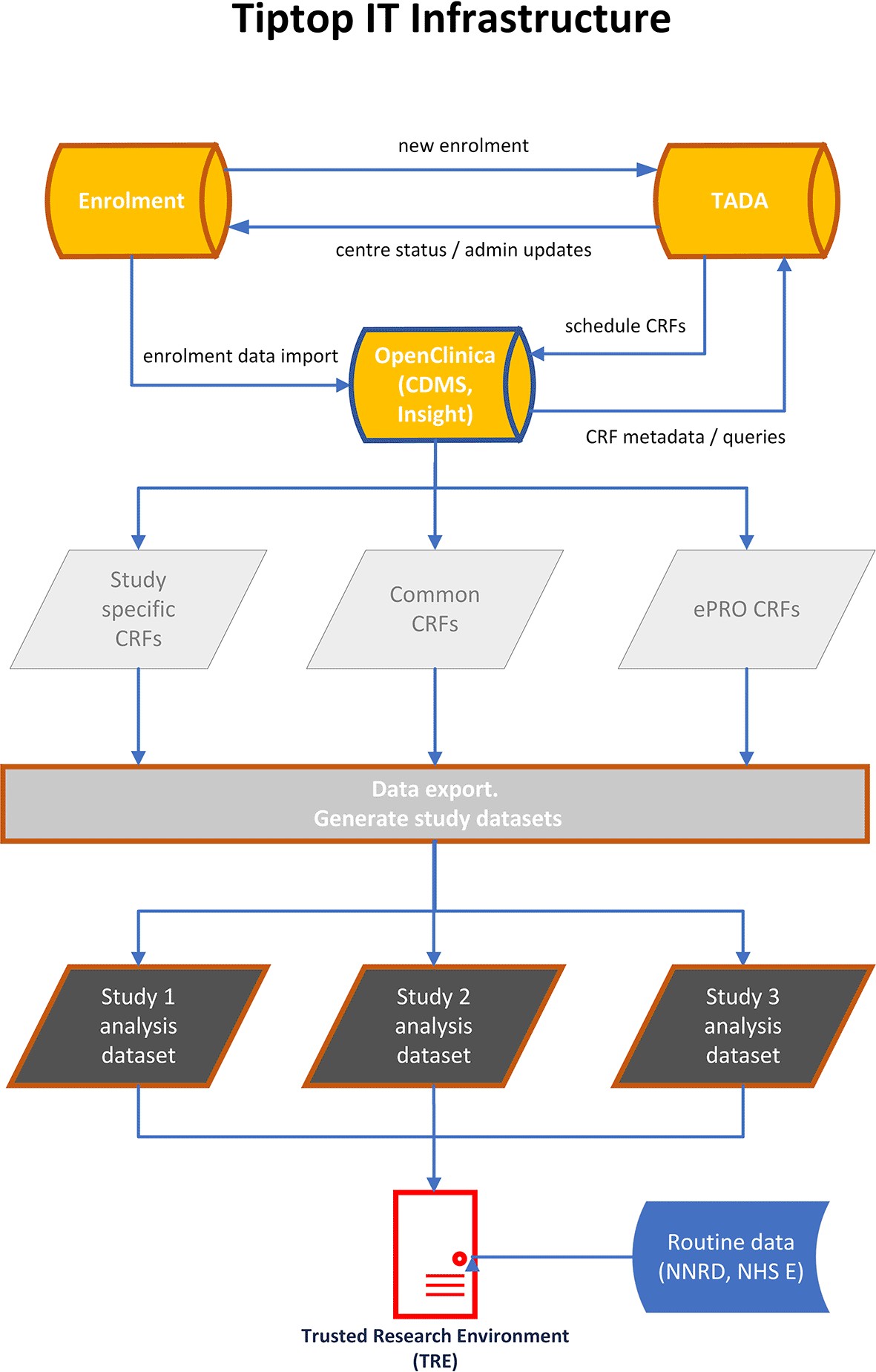
3.5.2 Enrolment Application
Based on the current NPEU CTU randomisation application (used in RECOVERY), this would be adapted to allow site staff to enrol new participants into a study. When participants were initially registered, each would be assigned a unique platform ID (UID), along with a unique study ID (see Figure 5).
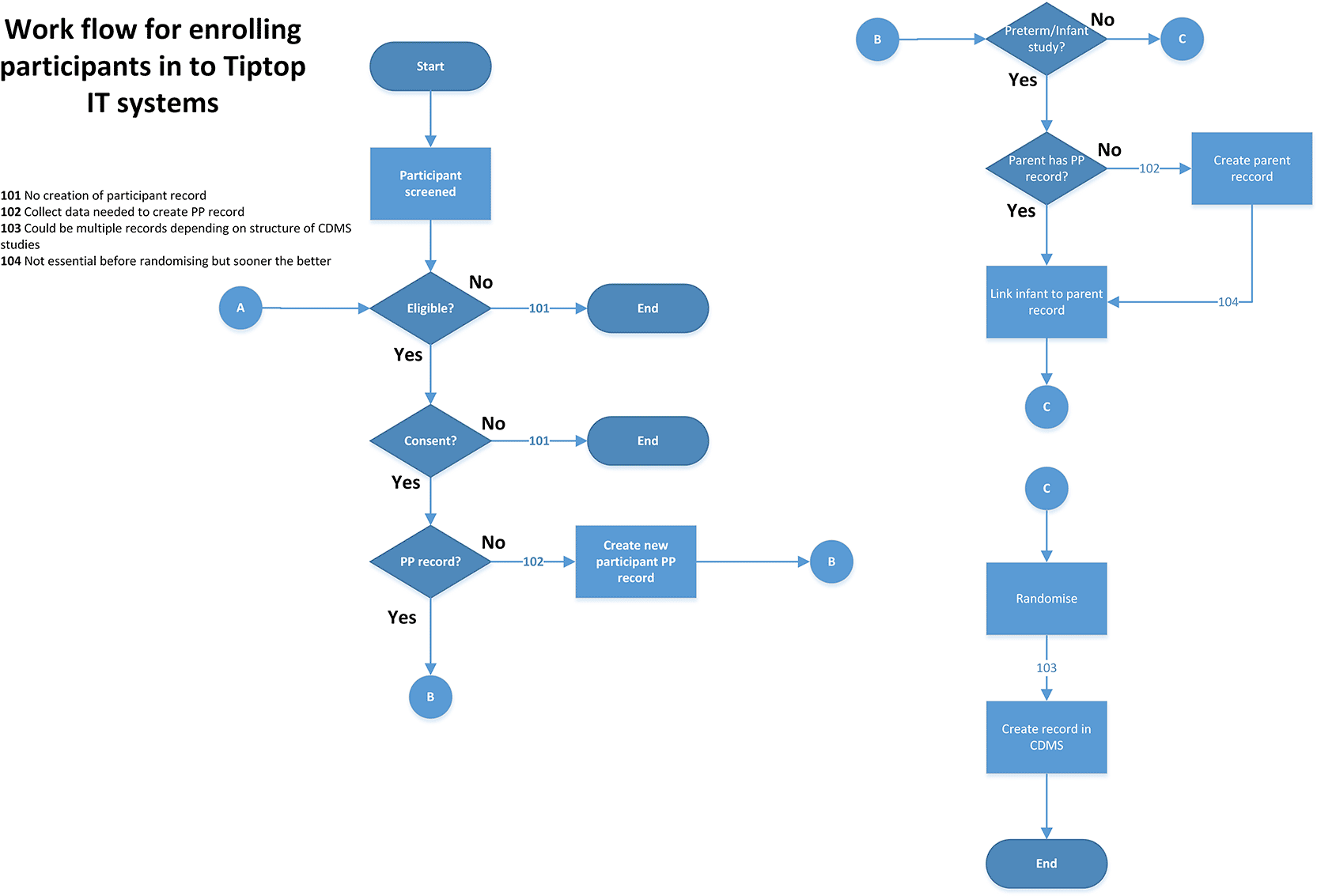
3.5.3 Trial Administration Database Application (TADA)
TADA is a bespoke trial administration application that is used to support the RECOVERY trial. An adapted version of TADA was to be used to support the administrative processes of running the platform (generally by CTU coordinating centre staff ).
TADA includes a data store of any non-clinical data relating to participants (identifiers, contact details, etc.) that would be needed to operate the trials in the platform, for example, for the purposes of contacting participants for follow-up or other data-gathering. Women and babies were both considered to be participants at the same level, with their interrelationships being stored as well.
TADA also holds a data store of all sites participating in any of the comparisons, and details of the research staff at those sites. The application was to be used to track staff training, delegation logs and involvement with participants (e.g. staff performing randomisations or taking informed consent).
Finally, TADA would be used to provide management-level reporting concerning recruitment progress, data-collection progress and other reports that might be deemed necessary.
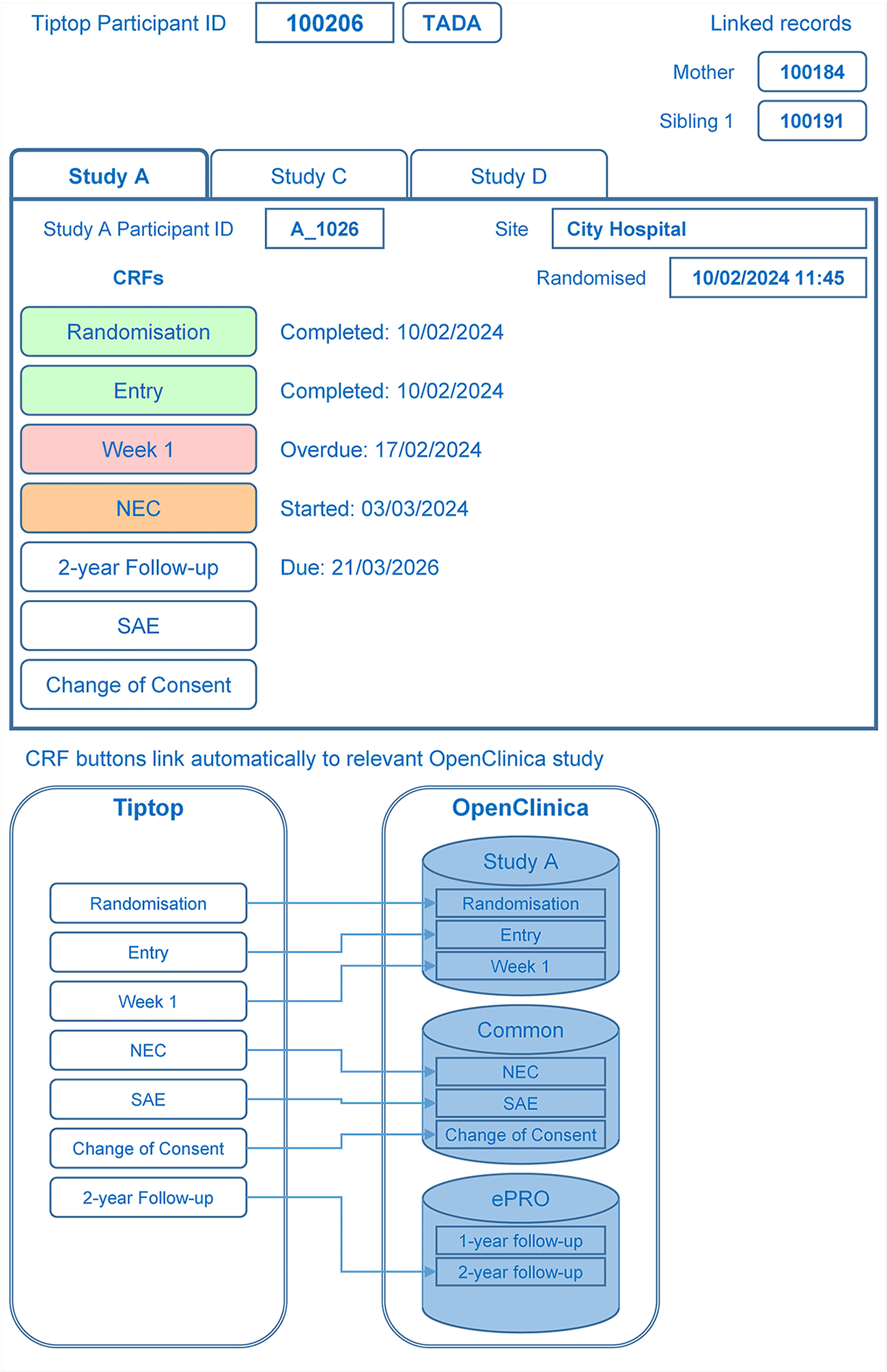
3.5.4 Bespoke data collection – OpenClinica
The OpenClinica CDMS would be used to collect and manage study-specific clinical data. To reduce the data entry burden of site staff, Case Report Forms (CRFs) would be shared across multiple studies; for example, in the case of two studies with a parent questionnaire at 2 years, the parent would only be required to complete one questionnaire.
To allow participants to enrol into multiple studies, consideration was taken on how the OpenClinica CDMS would be configured; the three options considered were:
1. One OpenClinica study to include all participant records across all studies on the platform;
2. One OpenClinica study to contain all CRFs for each platform study;
3. One OpenClinica study for each platform study containing study-specific CRFs, one OpenClinica study containing clinical CRFs, and another containing participant-reported questionnaires that are both shared across multiple platform studies.
Option 1 users would require access to only one OpenClinica study, but as more studies are added the performance of the CDMS would be affected, especially as functionality to archive participant data for completed studies does not currently exist within the system. Over time, the OpenClinica study could become unmanageable for all users (site staff and coordinating team); option 1 was rejected.
The benefit of option 2 is that all study data are contained within one OpenClinica study, so simplifying the process for uploading data to the Trusted Research Environment (TRE). However, if a participant were enrolled into multiple studies, an undesirable effect would be an increase in the data entry burden for site staff, as data would not be shared between OpenClinica studies, making option 2 unfeasible.
The preferred approach was option 3. It allows for the sharing of common clinical and participant-reported data across multiple studies, but also silos the study-specific data. It would require users to switch between OpenClinica studies, but this is a current requirement if users work across multiple studies. Preparing datasets for the Trusted Research Environment (TRE) would require processes for linking data from the multiple studies, but this was preferable to increasing the data entry burden on the users.
Having decided to store data for one participant in CRFs in multiple OpenClinica studies, it was necessary to create a system whereby users could easily navigate to those CRFs without having to work out which OpenClinica study contained which CRF. A portal was therefore designed to enable this feature, and users would access this portal within the Enrolment Application. It would show which study(ies) the participant had been enrolled in and for each study would display a tab showing buttons providing links to each CRF in the relevant OpenClinica study (see Figure 6).
3.5.5 Routine data maximisation over bespoke data
After determining the relevant information needed to answer these important clinical research questions, we identified the most reliable and robust national data sources needed to extract this information to reduce the burden of the manual collection process ( Table 3). We considered many inputs from experts and previous users of the concerned datasets to inform the final decision on whether each data item should be obtained manually or from national routinely collected data sources. Tables 4–6 show the final agreement for all the outcomes and data sources for the proposed comparisons.
Additionally, routine data requests for clinical research can be a long and costly process that needed to be simplified and streamlined to reduce research waste and excessive costs. Therefore, the identification of common outcomes for the three studies, has enabled us to minimise the data collection burden by planning to request data for multiple questions in one single application. Moreover, as the three trial populations inevitably cannot completely overlap, multiple releases of data were planned to cover the total combined population.
3.5.6 Trusted Research Environment/Data storage
To ensure the highest security standards were followed, to minimise the risk of data breaches, we planned to use a TRE. This is a highly secure digital environment that provides remote access to information and analytical tools in a single place. Holding the trial platform data in a single safe place enables the most efficient way to collect, process and link data for current and future trials. We discussed specifications and costs with the TRE provider at the University of Nottingham and obtained a tailored solution for our needs. In particular, we planned to store the data in the TRE for the whole length of the project, except for the first year where trial setup was needed. Due to the complexity of combining three trials in one platform (and potentially adding further ones in the future), access to the TRE was also planned to be continuous over the study length to allow for different users to process and analyse data in parallel for different studies and with no interruptions. At the end of the trial, routine data from each data provider was planned to be safely destroyed in accordance with each data provider agreement. The final pseudonymised dataset and the list of identifiers (separated from the dataset) were planned to be archived and retained for a period of 7 years (University of Nottingham policy) before being securely destroyed. As per Clinical Trials regulations, clinical trial data should be kept for a minimum of 5 years; however, academic policies might require a longer time.
Figure 7 shows the flow of data planned to be obtained from different data providers within the UK reaching the final secure destination in the University of Nottingham TRE, where linkage and processing were planned to be carried out for the creation of the final datasets for the analyses. Data managers, statisticians and health economists were planned to access the TRE remotely to conduct processing and analysis.
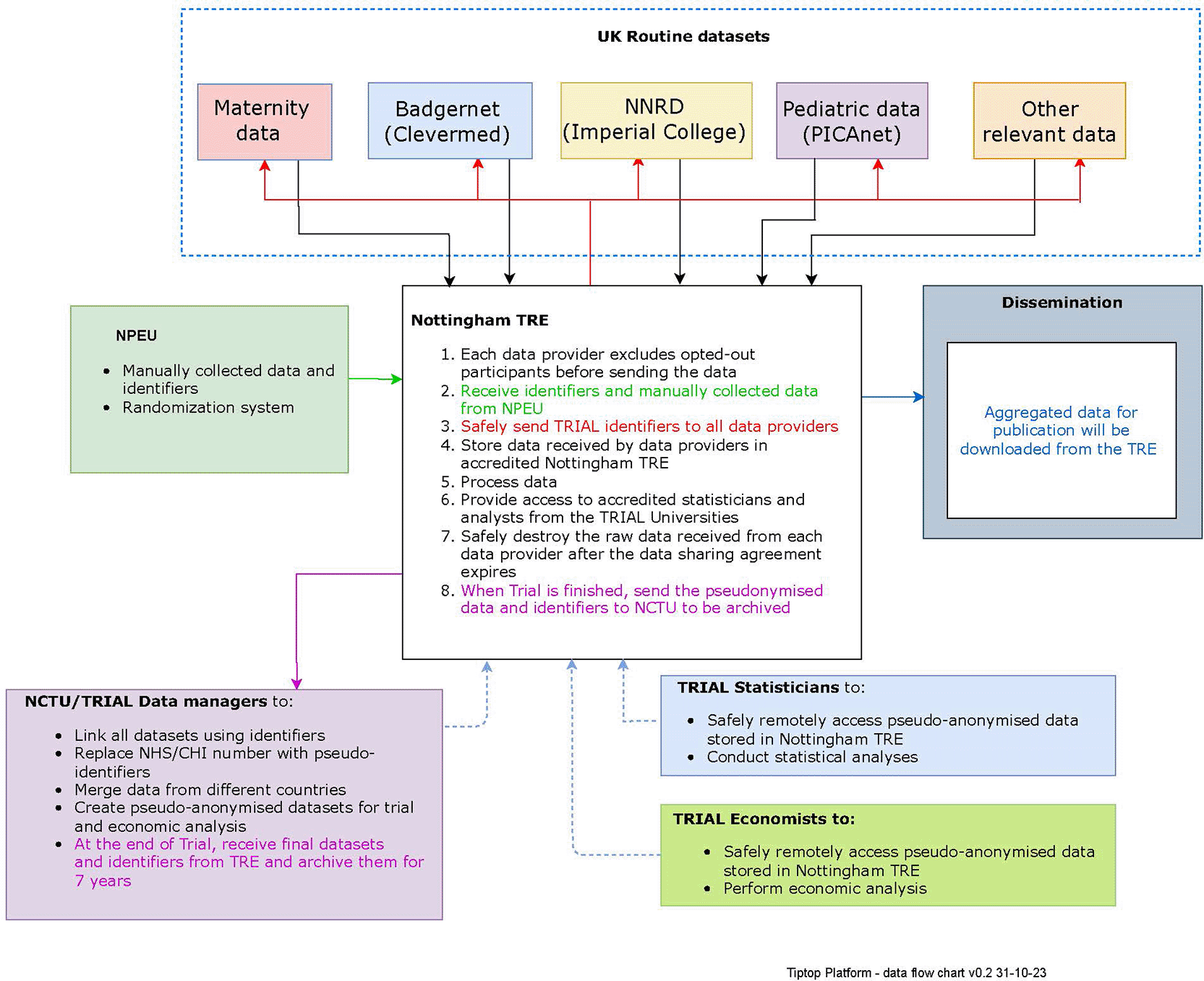
We planned to collect single identifiers manually on sites to identify each trial population and allow for the extraction and linkage of data obtained from several routine data sources. The three main identifiers, NHS number, date of birth and postcode were then planned to be sent to each data provider for them to extract the population and their records after selecting specific datasets, fields and time windows in accordance with each study recruitment period. The data processing techniques and the creation of the final datasets for analyses were planned to be developed following discussions with statisticians and health economists to allow for any specific requirements to meet the relevant adopted methodology. Overall, we planned three releases of data from each data provider to allow for quality and completeness checking. Interim releases of data also allow for the development of methodologies and algorithms to clean, link and analyse the data well ahead of the final analysis to account for any potential need to amend data requests and methods of analysis due to unpredicted data quality and completeness issues.
3.5.7 Information Governance for routine data
As consent was sought from each participant, the Confidentiality Advisory Group (CAG) approval for England and Wales and the Public Benefit and Privacy Panel for Health and Social Care (HSC-PBPP) approval for Scotland was not needed; and Information Governance (IG) approval was planned to be obtained from each data provider, as standard process for data request application review.
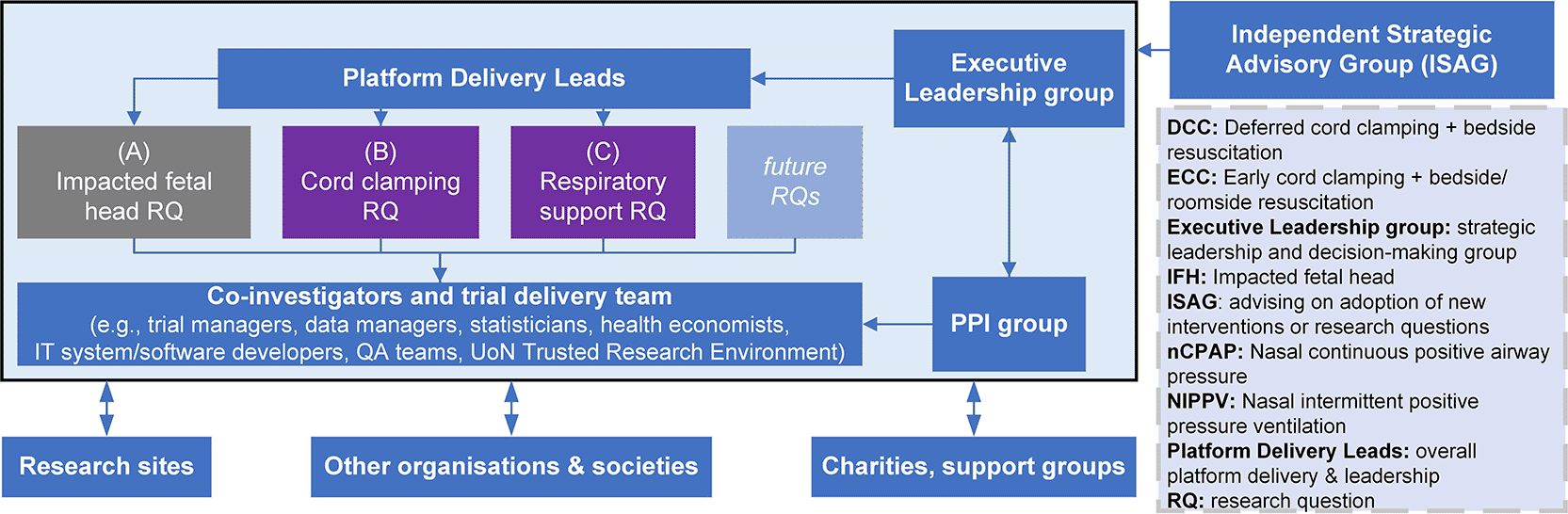
The aim was to refine the initial research questions and develop a high-quality grant application to the November 2023 commissioned call. All of the work described above, i.e., assessment of HTA readiness of proposed interventions, co-design with parents (assembling a diverse and dynamic PPI group with broad coverage) and healthcare professionals (facilitated through focus groups), consultation with governance teams, creation of oversight committees, methodological work (e.g., simulation studies, assessment of feasibility of recruitment) and IT systems development (modelled on the RECOVERY platform trial), contributed in some way to the proposed platform trial. Of course, this took exceptional leadership and organisation, included many meetings (face-to-face and hybrid) and several iterations! The leadership structure and organisation was complex but worked well (see Figure 8 for the overall structure and Table 1 to see how we organised ourselves overall and through working ‘subgroups’).
The Tiptop perinatal platform proposed addressed three research comparisons to prevent maternal morbidity, improve infant survival and neurodevelopment and prevent hypoxia, infant brain injury and chronic lung disease (see Figure 9). Namely:
(A) interventions to manage an impacted fetal head. Evaluating two techniques for managing an impacted fetal head at caesarean birth when labour has resulted in the head becoming impacted in the pelvis: the fetal pillow, and the standard approach, the vaginal push technique. [Note other techniques could have been added as they evolved.]
(B) timing of umbilical cord clamping when resuscitating very preterm babies. In babies born before 32 weeks’ gestation who need resuscitation, evaluating deferring clamping the umbilical cord for between 3 and 10 minutes compared to early clamping in the first minute from birth.
(C) modes of primary respiratory support for very preterm babies. In babies born before 32 weeks’ gestation who do not need resuscitating (i.e., ineligible for early clamping), evaluating the use of early nasal Continuous Positive Airway Pressure (nCPAP) compared to Non-Invasive Positive Pressure Ventilation (NIPPV).
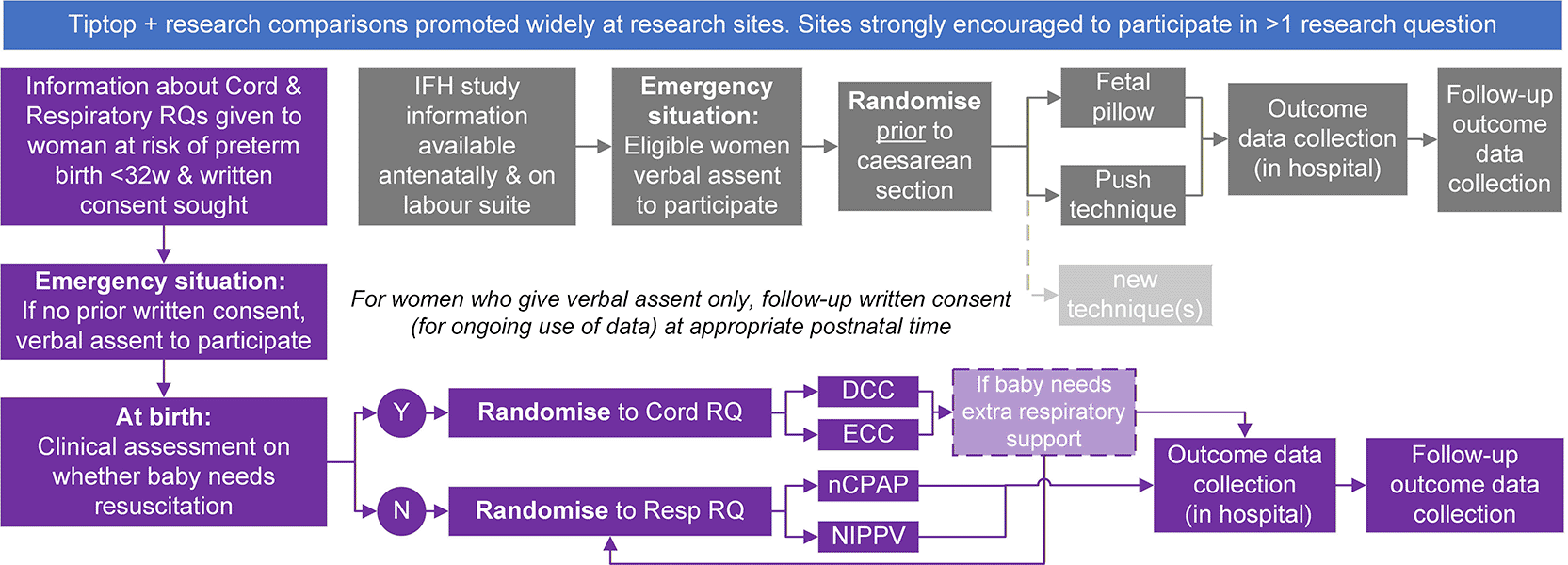
Reducing maternal and neonatal mortality and morbidity is a key Government priority in the UK.1–3 Problems that occur during pregnancy, birth and the newborn period are intrinsically linked and may result in death or survival with significant life-changing morbidity for the mother and baby. By design, by uncoupling woman and baby, randomised controlled trials in this field are disjointed and inefficient. This results in inadequacies in addressing the needs of pregnant women, parents, patients and the public, thus impacting the speed of delivery of evidence-based interventions. Such inefficiencies also fail to improve outcomes for women and babies, and fail to maximise the efficiency of the approach, consent, and data collection aspects of trial delivery, which are burdensome and duplicative, fuelling research waste. The perinatal period is a unique scenario during which interventions can be aimed at pregnant women antenatally, women and their babies during and following birth. We need to address this by designing trials that fully account for the mother-baby dyad and harness the power of novel trial designs, allowing the evaluation of the potential interventions efficiently. Innovation and the ambition to address these issues and responsibly deliver robust and timely randomised controlled trial evidence for this population are paramount.
We have proposed a platform infrastructure capable of hosting trials in which multiple research questions could be simultaneously addressed. This is achieved by streamlining processes and linking data across pregnant and birthing women and their babies. This report details the work undertaken to explore the concept of delivering a ‘perinatal platform’ in terms of organisation, governance, methodology, IT, with clinical and PPI perspectives front and centre. Three research questions were proposed on which to launch the platform, and submitted to the NIHR HTA as a Stage 1 application in November 2023, but this was unsuccessful.
4.1.1 The concept
Developing the concept and overall aim of the research design proved challenging for several reasons. Initial discussions centred around whether the platform should focus on a single disease area within the broader field of perinatology (e.g., improving outcomes for preterm babies), or whether greater efficiencies could be found by widening the scope to include research questions relevant to the population of mothers and their babies. Given that the health of and outcomes for the pregnant and then birthing woman and her baby are inextricably linked, and that a step-change in the delivery of efficient research in this field was considered essential, it became clear that the design of the platform had to incorporate research questions relevant to both. The subsequent discussions and development work then centred around the fact that we were designing a platform of trials applicable to a defined population rather than a particular disease area as is more commonly seen.
Nomenclature then affected how we communicated our intended research plan among ourselves as a large study team coming from a range of backgrounds. Were we planning a platform trial or a trials platform? How would this be different to an adaptive design? And what were the implications of each design? There is no clear agreement in the research community on what comprises a “platform” or an “adaptive trial”. Communicating this to clinicians, PPIE and the funder would also require careful attention.
Delivering this concept, although feasible and desirable, would require careful planning of the required infrastructure, and funding to put this in place, with economic efficiencies not evident until the platform was up and running. This was arguably the biggest challenge of all.
4.1.2 Leadership, governance and sponsorship
The traditional model of standalone trials is pervasive in clinical research. For all its inefficiencies, one advantage is that its ownership is clearer. Research projects require a named Chief Investigator and sponsor institution. By contrast, a platform of research which can change over time is always more expansive and complicated. Asking a potential lead investigator to be a co-investigator within a wider team limits their intellectual, academic and financial rewards. Asking an institution to sponsor a study in which large components are lead from elsewhere is similarly problematic.
In a similar vein, the ambition to work across numerous clinical trials units with complementary skills and experience, to provide a cross-institution “virtual trials unit” would be challenging to operationalise and expensive to deliver. The current devolved system of a network of academic CTUs does not lend itself easily to such an endeavour. Using staff interchangeably across ‘sister sites’ is common in large corporations. It is less easy to achieve this when the funding is awarded directly to the sites, each of which have their own financial interests and human resource structures. To give a concrete example, if institutions A and B both receive funding to cover a 1.5WTE study manager but subsequent changes require an unequal split, it is less easy to transfer funding or staff between the two institutions. This matters more on platform designs in which adaptations are to be expected.
Sponsorship was a potential issue, although a way forward was agreed. The proposed platform trial funding application needed a named sponsor, whereas sponsors are usually responsible for an individual trial rather than a platform of several trials. This can create tensions over who assumes responsibility as sponsor for the overarching platform. Again, to give a concrete example, our platform was set up to start with three trials addressing separate (but linked) research questions, anticipating these would be added to or removed in the future. Moreover, the intention to make the platform open and inclusive to researchers from other institutions meant that research questions could originate from institutions external to the platform’s host institutions. Indeed, platform trial adaptations may mean that all trials proposed by the host institution may be discontinued. Not only does this take the host institution into the difficult territory of sponsoring studies that they have less expertise in, but there is also the politically difficult issue of ownership of intellectual property. Whilst our team and institutions were open and collaborative, and some scenarios were likely only hypothetical, there were some difficult discussions needed to reach resolutions. This vital step must not be avoided by any team planning to take the same approach.
Agreeing/harmonising a set of working guidelines or standard operating procedures was identified as an important factor to enable the delivery of the platform, but one we believed to be low risk. There was experience in the team in running trials to a mix of internal and external SOPs for studies with an external sponsor, and for where study-specific SOPs were needed. Adequate documentation, training and awareness would be required. Nonetheless, time and thought would be needed to agree on the SOPs or working guidelines, filling in the gaps necessitated by a complex platform design with study-specific SOPs and training.
4.1.3 Methodology
We were able to identify important and common outcomes relating to trial interventions delivered across the perinatal period and to be evaluated within the platform. Multiple routine data collection sources were identified to minimise bespoke data collection requirements. Further work is required to assess and enhance the reliability of the data contained in these sources. Challenges were anticipated in the timings of routine data downloads and the corresponding timings of multiple interim analyses. A key strength of the platform infrastructure would be to streamline the collection of long-term neurodevelopment and health economic outcomes across trials on the platform.
Adaptive design methodology was employed to ensure a cost-effective platform design. This would allow prespecified trial adaptations to balance the delivery of robust trial evidence and prioritise funding for ongoing recruitment to research questions that showed promise of delivering useful information. Detailed statistical simulations were undertaken to assess design performance characteristics under a variety of scenarios. Input from the clinical investigators was required to finalise interim decision rules (e.g., stopping guidelines). Communicating the complex statistical concepts to non-statisticians, and the wide range of scenarios required for the multiple research questions was challenging, although achievable.
4.1.4 IT solutions
Using our existing systems as a starting point, the same systems used for the RECOVERY trial, we explored the development of an IT solution that would allow for the complexities around the overlapping research questions and target populations. This solution would integrate data across research questions on the platform in terms of participant enrolment, trial administration, the Clinical Data Management System, and allow for the incorporation of routinely collected data. The data would then feed into a Trusted Research Environment to allow access and analysis by people across different institutions.
The bespoke Enrolment Application and Trial Administration Database Application could be adapted to appropriately incorporate the specifications required. However, compromises would be required when using an ‘off-the-shelf’ CDMS. The proposed system would make use of OpenClinica (the system used for all NPEU CTU trials) to minimise any cost outlay. OpenClinica is structured so that all data for one participant in a study is stored within a single OpenClinica study. However, the platform would require data for one participant to be stored in several different OpenClinica studies. Users would therefore need to be given accounts with permission to access multiple studies and a separate portal (within the Enrolment Application) would require development to allow easy access to those different studies.
Further complications arise because, unlike in standalone trials, data may be linked to more than one participant. For example, a mother’s 2-year follow-up data could be required for analysis in different studies involving the mother and their infant(s). The entry of this data would be managed via the portal; this would provide a link to a single CRF that could be part of the mother’s or infant’s record depending on which participant was first recruited to the platform. This data would then need to be included in the dataset for each relevant study.
Lastly, the platform was designed to incorporate routinely collected data in a bid to reduce both inefficiencies and burden and on recruiting sites. The advantages of this are self-evident, especially in the present time when NHS maternity care resources are limited, but its key downside is the upfront investment in developing the system. This inevitably led to a platform whose efficiencies came at a cost that would only be recouped after several trials, and thereby less competitive when judged on its original three research questions.
4.1.5 Parent, Public and Healthcare Professionals’ views
Parents and members of the public were at the heart of our plans throughout, and we learnt a great deal from them when considering the design and running of a future perinatal platform trial. Working closely with the PPI group, the value of parent-led PPI initiatives has been highlighted – this is not something we had experience of previously, but would certainly implement in future trials as a way of including a much wider group of parental voices. We have communicated with the PPI group in ‘non-traditional research’ ways, for example by using a WhatsApp group, which has led to a closely knit group of parents forming. They have expressed that they have enjoyed the process and would like to be involved in developing future research studies. Given the sensitive nature of discussions relating to preterm and complicated births, we have learnt the importance of creating a safe space for discussions, where everyone’s views are valid and, when triggering conversations do occur, how we can support one another.
The online discussion groups that were held with multidisciplinary healthcare professionals were greatly informative, though would have been enhanced by having more time for deeper discussions. However, this was difficult to balance given the complexities of the platform trial design, the number of research questions and busy healthcare professionals volunteering their time freely.
Although full validation of the perinatal platform trial through implementation with an intervention comparison has not been performed, several components of the proposed infrastructure were operationalised and tested during the accelerator phase.
Statistical simulations were conducted to explore operating characteristics of an adaptive perinatal platform design, including power, type I error control, and the feasibility of adding and dropping comparisons. These simulations informed the proposed analytical framework and confirmed methodological feasibility within realistic recruitment projections for UK perinatal populations.
The governance model was developed through iterative consultation with multi-stakeholder groups, including sponsors, clinical trials units, clinicians, methodologists, parents and national charities. Draft processes for intervention selection, prioritisation, and oversight were planned (as structured workshops and consensus exercises); not all were piloted.
The IT and data flow framework was designed in collaboration with multiple clinical trials units to enable shared data capture and harmonised outcomes across maternal and neonatal components. Prototype data structures and workflow maps were developed and reviewed across institutions to assess interoperability and regulatory compliance. Many of these were adapted from a highly successful large international platform trial (RECOVERY), accounting for the mother-infant interrelationship. Participant data would be securely hosted within the well-established Trusted Research Environment at the University of Nottingham (a secure haven for sensitive/confidential data).
Public and patient involvement structures were implemented during the development phase, demonstrating feasibility of embedding the voices of parents and people with lived experience within platform prioritisation and design processes.
Ultimately, the methodological framework drew upon established adaptive and platform trial models successfully implemented in other clinical areas (e.g., multi-arm multi-stage and adaptive platform trials), adapted specifically to address the unique interdependence of maternal and neonatal outcomes.
Together, these applications demonstrate the practical feasibility of the proposed blueprint, even though full platform implementation remains to be realised.
4.2.1 Reflections
The use of platform trials remains uncommon despite their potential efficiencies. The difficulty in operationalising these (described above), along with a lack of expertise, means conventional clinical trials tend to be conducted using individual processes. Platform trials have been successfully deployed in a number of settings, most frequently oncology and infectious diseases, although typically with less expansive designs than that outlined here.42
Two notable examples of large, multi-factorial adaptive platform trials are the international REMAP-CAP and RECOVERY trials which faced many of the same issues outlined here – multiple sponsors/stakeholders overseeing multiple comparisons, and each of which could theoretically add or remove interventions. Many of the challenges seen in these studies are summarised in a reflection paper on the EU-PEARL platform.43 Importantly, the set-up and sustainability of all three platforms was made possible by substantial funding and national trial infrastructure/system changes, which is clearly an exception to the norm.
The breadth of the network and of the planned work presented many challenges.
Collaboration across several CTUs can provide the necessary complementary expertise required for innovation in terms of shared learning, training and shadowing opportunities, efficiencies in maximising a limited pool of expertise and minimising competing studies, improved communications, and most importantly, offering pregnant women and parents all possible research options available in a coherent, transparent and equitable manner. However, whilst institutions and individual researchers are primarily judged by research income, competition inevitably makes such endeavours complicated.
Governance and legal aspects have yet to evolve sufficiently to reassure host institutions regarding risk management across multiple studies and institutions. Despite this, the sponsor teams at the two main institutions were keen to develop a complex, agile sponsorship framework implementing lessons learned from COVID-19 and other platform studies.
It was imperative that the proposed platform should allow adaptations beyond adding or removing interventions: we needed an agile infrastructure within which new trials could be added. Developing an infrastructure that was adequately futureproofed required an upfront investment, and this offset many of the efficiencies that platform trials promise over three standalone trials.
None of the challenges that we were faced with were insurmountable. However, the ambitious nature of the Tiptop perinatal platform required substantial start-up investment, which would only confer benefits to the women, their families, the NHS and wider society, downstream. These benefits might only be harvested in years to come e.g., following 2-year follow-up of infants and beyond in terms of educational attainment. Reducing brain injury in babies, for example, would reap sufficient rewards both directly to individuals and indirectly to society, but one could argue that the risk of putting all of one’s eggs in one basket was one leap of faith too far in the present economic climate (‘jam tomorrow’).
Nevertheless, to reiterate, this project has demonstrated the practical feasibility of the proposed blueprint, even though full platform implementation remains to be realised. We believe we have created a road map for the conduct of future more integrated, streamlined and inclusive perinatal trials.
Ethics approval was not required for this project, since it focussed on the development of a future clinical trial - no human participants were involved. Parents provided their views and experiences as part of our extensive public involvement activities (see Section 1.3.3) and offered their verbal consent prior to talking to us, but formal, written consent was not required, since this was not a clinical research study.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)