Keywords
Chronic Pain, Opioids, Tapering, Primary Care, Cluster Randomised Controlled Trial, Process Evaluation, Health Economics
Chronic Pain, Opioids, Tapering, Primary Care, Cluster Randomised Controlled Trial, Process Evaluation, Health Economics
 How to cite: Ashworth J, Cornwall N, Harrisson SA et al. Evaluating a primary care pharmacist-led intervention to reduce opioid use for persistent non-cancer pain: the PROMPPT cluster randomised controlled trial protocol [version 1; peer review: 1 approved with reservations, 1 not approved]. NIHR Open Res 2025, 5:54 (https://doi.org/10.3310/nihropenres.13961.1)
First published: 03 Jul 2025, 5:54 (https://doi.org/10.3310/nihropenres.13961.1)
Latest published: 19 May 2026, 5:54 (https://doi.org/10.3310/nihropenres.13961.3)
How to cite: Ashworth J, Cornwall N, Harrisson SA et al. Evaluating a primary care pharmacist-led intervention to reduce opioid use for persistent non-cancer pain: the PROMPPT cluster randomised controlled trial protocol [version 1; peer review: 1 approved with reservations, 1 not approved]. NIHR Open Res 2025, 5:54 (https://doi.org/10.3310/nihropenres.13961.1)
First published: 03 Jul 2025, 5:54 (https://doi.org/10.3310/nihropenres.13961.1)
Latest published: 19 May 2026, 5:54 (https://doi.org/10.3310/nihropenres.13961.3)
Persistent pain, defined as long-term pain not caused by cancer, is a common problem affecting an estimated one-third to one-half of adults in the United Kingdom (UK)1 and around one in five adults in the United States (US)2. Opioids are commonly prescribed for persistent pain3,4, despite a lack of evidence for their long-term efficacy and the risk of serious harm5–7. A recent review estimated that almost one-third (31%) of patients with persistent pain are prescribed an opioid4 and reducing inappropriate opioid use is a key healthcare priority in many countries8. Most long-term opioid prescribing for persistent pain occurs in primary care and best practice guidelines recommend regular review of patients on opioids and gradual tapering of opioids (opioid deprescribing) if treatment goals are not met8. However, research suggests that opioid deprescribing is viewed as challenging and time-consuming by healthcare professionals9 and, with limited time available in UK general practitioner (GP) appointments, approaches to reviewing opioids in primary care are variable and often ‘light- touch’10. Furthermore, there is a lack of practical guidance on how to implement opioid deprescribing.
Systematic reviews have found limited evidence, often of low methodological quality, supporting interventions to reduce long-term opioid use and recommended further, larger randomised controlled trials (RCTs) of theoretically grounded behaviour change interventions to support opioid deprescribing in the context of persistent pain, which measure long-term follow-up and include a health economic evaluation11–13. Sandhu et al. evaluated a multicomponent community-based group intervention, incorporating individual tapering support, to reduce opioid use (iWOTCH14). They found that, among UK patients with persistent non-cancer pain, a group-based educational intervention significantly reduced opioid use compared with usual care without affecting perceived pain interference with daily activities14. However, less than half of participants randomised to the group intervention attended all sessions. Given the diversity of patients who are prescribed opioids for persistent pain, not all of whom will be able or willing to engage with a group intervention, a range of evidence-based strategies to reduce opioid use are likely to be needed15.
To address the need for primary care-based interventions to support opioid deprescribing in the context of persistent pain, we developed the PROMPPT intervention (Proactive clinical Review of patients taking Opioid Medicines long-term for persistent Pain led by clinical Pharmacists in primary care Teams). PROMPPT capitalises on an expansion in the number of clinical pharmacists working in UK general practices (practice pharmacists), who play an increasing role in managing patients on long-term medicines by conducting consultations with patients in GP surgeries16. In line with Medical Research Council (MRC) guidance on the development and evaluation of complex interventions, the PROMPPT intervention was co-designed with stakeholders17 (patients and healthcare professionals), using a person-based approach18, combined with best practice guidance and theory on behaviour change. Extensive intervention development work included interviews with patients, pharmacists and GPs, an online qualitative study and in-practice testing of prototype PROMPPT consultations19–21, followed by a non-randomised feasibility study22. At each stage, findings informed iterative refinement of the PROMPPT review and pharmacist training. We hypothesise that the PROMPPT intervention will increase the likelihood of patients with persistent pain reducing their opioid use, compared to usual care, and that this can be achieved without increasing pain and pain-related interference.
The current PROMPPT trial protocol (version 1.6 2 Apr 2024) is described below, in line with SPIRIT reporting guidelines23. Key amendments to the protocol since initial trial registration are highlighted in the text with comments in brackets and summarised in Table 1.
| Amendment number* | Protocol section | Original protocol | Amendment details† | Date | Rationale for amendment | |
|---|---|---|---|---|---|---|
| 2 | AM02 NSA 02 | 7.8 Long-term follow-up assessments 7.10 Withdrawal criteria | Individual clinical outcome data Where there are ambiguous or incomplete pain medicines use questionnaire entries that interfere with calculation of daily MED, a member of Keele CTU staff will try to contact participants by telephone to obtain the missing data. | Individual clinical outcome data Where there are ambiguous or incomplete pain medicines use questionnaire entries that interfere with calculation of daily MED, a member of Keele CTU staff will try to contact participants by telephone or in writing, to obtain the missing data. Withdrawal criteria Participants that have consented but advise that they do not take opioids, will be subsequently withdrawn from the study, due to not meeting the inclusion criteria. | November 2022 | To improve the completeness of data collection, and ensure participant eligibility if participants cannot be contacted by telephone, participants with missing or ambiguous questionnaire entries regarding their pain medicines questionnaire may also be contacted by letter. |
| 3 | AM03 NSA02 | 7.9 Process Evaluation (MOPP-2 Study) | Identification and recruitment of participants (MOPP-2) Patients Participants from clusters in the intervention arm who consented to being contacted about a future related research study and subsequently attend a PROMPPT review will be invited to participate in the process evaluation (MOPP-2 study). | Identification and recruitment of participants (MOPP-2) Patients Participants from clusters in the intervention arm who consented to being contacted about a future related research study and subsequently attend a PROMPPT review will be invited to participate in the process evaluation (MOPP-2 study). Reminders will be sent to non-responders at 2 weeks either by email or post dependent on patient preference. | January 2023 | As part of the process evaluation, participants who consented to further contact are sent a Process Evaluation (MOPP-2 study) recruitment pack including an Acceptability Questionnaire. The return rate for Acceptability Questionnaires was lower than expected and the original protocol did not allow for reminders, so this was added to the protocol and new reminder letters created. |
| 4 | AM04 SA01 | 7.8 Long-term follow-up assessments 7.10 Withdrawal criteria | Individual clinical outcome data Standard Keele CTU procedures will be followed to maximise follow-up at all timepoints (3, 6 and 12 months). This includes electronic / postal reminders for non-responders after 2 and 4 weeks and contact by telephone for minimum data collection (MDC) after 6 weeks. | Individual clinical outcome data Standard Keele CTU procedures will be followed to maximise follow-up at all timepoints (3, 6 and 12 months). Online participants receive a text alerting them to the email containing link to follow-up questionnaire. Non-responders receive a text and email reminder after one week and a postal questionnaire after 2weeks. Postal participants who do not respond are sent a reminder postcard after 2 weeks and a new postal questionnaire after 4 weeks Non-responders are telephoned up to 3 times for minimum data collection (MDC) after 6 weeks and receive text message prior to MDC calls. Postal MDC questionnaire sent to non-responders who are uncontactable by telephone. Those who participate in the study and return either a paper or online questionnaire at 12 months will be included in a prize draw for the opportunity to win one of three £100 prizes (e.g. Amazon voucher). Withdrawal criteria Participants opting to complete the questionnaire online will be advised, in the participant information leaflet, that they can stop completing the questionnaire at any time point, without giving a reason, but that answers entered up to that point will be kept unless they contact the study team to opt out of this. New patient-facing documents - New “Thank You” flyer with all postal questionnaires and “Thank You” banner in emails for online participants - New covering letter/email with 12-month follow-up questionnaire emphasising its importance and including optional entry to a free prize draw Revised order for follow-up questionnaires, with both co-primary outcomes at the start and fewer mandatory fields in the online version | April 2023 | In the internal pilot, 3 month follow-up rates were lower than expected, therefore a range of amendments to the protocol and associated patient-facing documents were made with the aim of improving follow-up data collection by: (1) improving follow-up questionnaire response rates, (2) allowing use of data from partially completed follow-up questionnaires unless participants opt out, and (3) improving minimum data collection (primary and key secondary outcomes) from non-responders. |
| 7 | AM07 NSA05 | 8.2 Planned recruitment rate | An internal pilot will check recruitment assumptions, and we will adjust the recruitment strategy accordingly. | July 2023: Internal pilot results recommended that practices with list sizes <5000 can also be recruited to the study and practices can invite up to 350 eligible patients per practice (sampled to reflect the distribution across opioid strength groups (weak, intermediate, strong) in the practice if there are an excess of 350 patients who meet the eligibility criteria for the study). | July 2023 | Following the internal pilot study, to facilitate recruitment of GP practices and the required number of participants within the planned timeframe, we adjusted the recruitment strategy to allow for smaller practices to be included whilst also raised the maximum number of eligible patients to be invited per practice. |
| 9 | AM09 NSA07 | 7.8 Long-term follow-up assessments | Practice-level outcome data We will extract and analyse aggregated data from the electronic medical records of participating practices on relevant prescriptions among adults aged ≥18 years who do not have a coded cancer diagnosis. | Practice-level outcome data We will extract and analyse pseudonymised data from the electronic medical records of participating practices on relevant prescriptions among adults aged ≥18 years, excluding those who have both a coded cancer diagnosis and a code for palliative care. | February 2024 | To exclude patients receiving end-of-life care for cancer and avoid unnecessarily excluding patients with a historical cancer diagnosis that was no longer relevant, and cancer survivors treated for chronic non-cancer pain from data on practice-level pain-related prescribing. |
| 10 | AM10 SA02 | 7.2 Patient identification 9.1 Data collection tools and source document identification 9.2 Data handling and record keeping | Eligible patients who have been identified and who have a mobile phone number that is able to receive SMS on the practice record will be sent an SMS text invitation from the general practice to take part in the research. The SMS will contain a secure online link URL, hosted on Keele University secure servers to an embedded patient information leaflet, consent form and baseline questionnaire for online completion. Participating GP practices will send an Excel file to the study team of those patients invited, via NHS.net secure email. A dedicated web-based Management Application will be developed and maintained by Keele University and data will be stored on Keele University secure servers and password protected. A study online survey will be developed and maintained on password protected Keele University servers for participants that choose to complete self-report data online. | Eligible patients who have been identified and who have a mobile phone number that is able to receive SMS on the practice record will be sent an SMS text invitation from the general practice to take part in the research. The SMS will contain a secure online link URL, hosted on Keele University managed storage services, to an embedded patient information leaflet, consent form and baseline questionnaire for online completion. Participating GP practices will send an Excel file to the study team of those patients invited, via secure transfer. A dedicated web-based Management Application will be developed and maintained by Keele University and data will be stored on Keele University managed storage services, held within the UK and protected by industry standard security tools. A study online survey will be developed and hosted on Keele University managed password protected storage services for participants that choose to complete self-report data online. | April 2024 | This amendment was required due to changes in how data collected to the trial is stored |
1. To determine, in patients prescribed opioids long-term (≥6 months) for persistent pain, whether providing the PROMPPT intervention (practice pharmacist-led primary care pain review and associated pharmacist training package) is more likely to reduce opioid use, without increasing pain/pain-related interference, at 12-month follow-up compared with usual primary care review.
2. To determine, in patients prescribed opioids long-term (≥6 months) for persistent pain, the differences, between treatment arms, in patient-reported clinical outcomes including pain, pain-related interference, use of opioid and non-opioid pain medicines, confidence to cope with pain, symptoms of depression and anxiety, presence and severity of opioid-related side-effects, and health-related quality of life at 3, 6 and 12-month follow-up.
3. To determine the differences between treatment arms in GP practice-level prescribing of opioids, non-opioid analgesics and other potentially sedating medicines commonly prescribed for patients with persistent pain at 12-month follow-up.
4. To estimate the cost-effectiveness of providing the PROMPPT intervention versus usual primary care review for patients prescribed opioids long-term (≥6months) for persistent pain.
5. To explore, through a mixed methods process evaluation, how PROMPPT was delivered and received, the quantity and quality of what was delivered (fidelity and dose), how PROMPPT impacted on any change in outcomes and how context affected implementation of PROMPPT and outcomes.
The PROMPPT trial is a pragmatic, multicentre, parallel two-arm cluster randomised controlled trial (RCT) with an internal pilot, linked health economic evaluation and mixed methods process evaluation. A cluster RCT design was chosen to (a) reduce treatment contamination risk between arms and (b) to facilitate recruitment, as practice level randomisation has previously been highlighted as a potential strategy for addressing the challenges of recruiting patients who may be apprehensive about changes to their opioid analgesics24,25.
The setting is UK primary care, specifically 38 general practices from across the West Midlands, East Midlands, Wessex, Thames Valley and South Midlands. Participating practices identify eligible patients from their electronic health records and invite them to participate in a questionnaire study (patient facing name: Management of Opioids & Persistent Pain (MOPP) Study). Practices are randomised (1:1) to either offer PROMPPT (intervention arm) or continue usual primary care arrangements for reviewing patients prescribed long-term opioids (control arm). The units of randomisation are the general practices, and the units of observation are MOPP study participants, who receive the care to which their practice is randomised. Participant flow through the trial is shown in Figure 1.
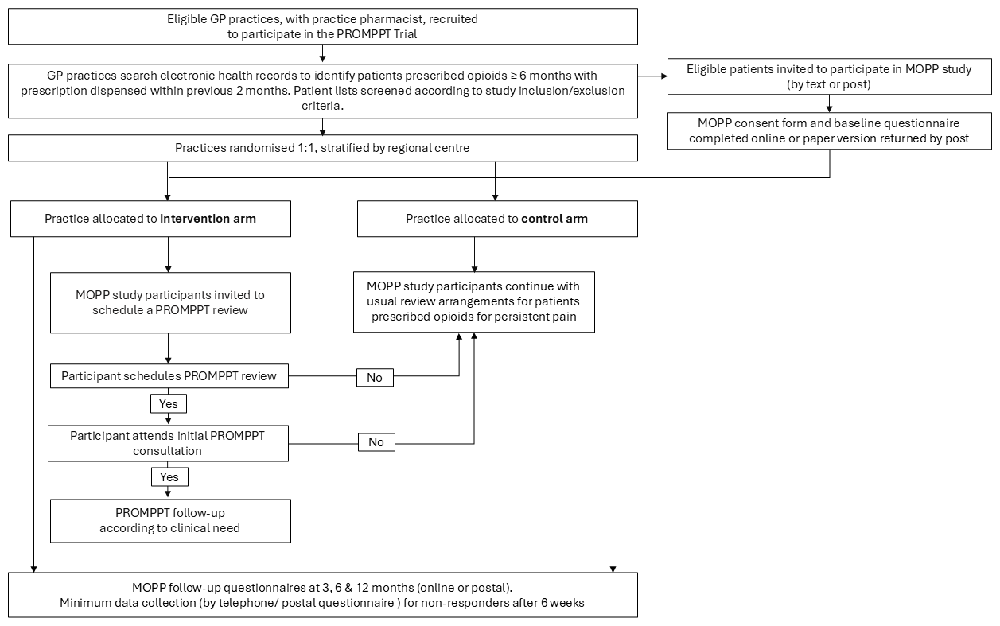
To minimise the risk of post-randomisation selection bias26, identification of eligible potential participants is completed prior to practice randomisation. In addition, practices are requested to provide pseudonymised demographic data for patients invited (including National Health Service (NHS) number, age, sex, and opioid strength grouping (defined below)) to the research team to allow any evidence of selection bias to be assessed. To reduce the risk of participation bias, all potential participants receive identical information inviting them to participate in the MOPP study and all patient participants complete identical MOPP study questionnaires at baseline and all follow-up time points, regardless of which treatment arm their practice is randomised to.
Ethical approval was obtained from North East, Newcastle & North Tyneside 2 Research Ethics Committee (REC) (Ref:22/NE/0044). Following initial approval, the REC has been informed of all substantial changes to the management of the trial.
General practices are eligible to participate in this trial if they use either the EMIS Web (Optum: https://www.emishealth.com/products/emis-web) or SystmOne (TPP: https://tpp-uk.com/products/) electronic health record (EHR) system and have at least one practice pharmacist, who is an Independent Prescriber and consults with patients face-to-face and/or remotely, working within the practice for at least one session per week. Initially practices were also required to have at least 5000 registered patients but from July 2023, practices with <5000 registered patients were also included (Amendment 7, see Table 1).
Recruitment of general practices is facilitated by National Institute for Health and Care Research (NIHR) Clinical Research Networks (CRNs) in the West Midlands, East Midlands, Wessex and Thames Valley & South Midlands to identify eligible general practices with a range of patient list size and setting (urban, semi-urban, and rural). Members of the research team meet with a GP, practice pharmacist and practice manager (face-to-face or remotely) to explain the study and describe the study requirements. GP practices are recruited in batches until the required number of participants is recruited. Informed written consent for practices to participate is provided by the authorised person in each practice, as a guardian for patients in their care, following agreement with the practice team to provide either the PROMPPT intervention or usual care (dependent on random allocation) for the period of the trial and to participate in the process evaluation.
Adult patients aged 18 years and over, registered at a participating practice, and prescribed any opioid analgesic (defined as any opioid or opioid/paracetamol combination analgesic from sections 4.7.2 and 4.7.1 British National Formulary (BNF))27 for persistent non-cancer pain for at least 6 months, with at least one prescription issued within the previous 2 months.
Patients being treated for acute pain (self-limiting pain, for example after injury or surgery), pain associated with cancer and patients with terminal illness (life expectancy <6m),
vulnerable patients (as assessed by general practices e.g. severe mental illness, learning difficulties, dementia),
patients currently receiving treatment for substance misuse.
Participating practices search their electronic health record systems, using a Search and Report, designed by a health informatics specialist from Keele Clinical Trials Unit (CTU), to systematically identify adults prescribed opioid-containing pain medicines for 6 months or longer (with at least one prescription dispensed in the last two months), grouped according to the strength of opioid medicine (weak, intermediate, strong, see Table 2), based on a published categorisation for prescribed analgesics in primary care28. Practices then screen patients identified according to study inclusion and exclusion criteria, excluding ineligible patients.
* Hierarchy of analgesic potency arising from a consensus study of UK general practitioners28
To avoid individual practices being overrepresented in the overall study population, the maximum number of patients invited to participate per practice is capped. Initially a maximum of 250 eligible patients per practice were invited, but the cap was increased to 350 from July 2023 to facilitate recruitment within the planned timeframe (Amendment 7, see Table 1). Where practices identify an excess of potentially eligible patients, a representative sample (randomly sampled by Keele CTU staff to reflect the distribution across the opioid strength groups (weak, intermediate, strong) in the practice overall) is invited by the GP practice.
Using general practices’ existing systems for bulk text messaging, potentially eligible patients with a mobile phone number on the practice record are sent a text message from their practice inviting them to participate in the Management of Opioids & Persistent Pain (MOPP) Study. The text message contains a secure online link to a web page, hosted by Keele University, with an embedded participant information leaflet, consent form and baseline questionnaire for online completion. A reminder text is sent after 2 to 3 days and includes information on how to contact Keele CTU for assistance completing the online questionnaire, or to request a postal version of the information leaflet, consent form and questionnaire. Potentially eligible patients who cannot be invited by text are sent a postal invitation pack via Docmail, a secure remote document compilation, print and mailing solution. To facilitate inclusion of patients, Keele CTU offer telephone support for questionnaire completion and reassurance that family members can assist; and for patients whose first language is not English, questionnaire completion can also be facilitated online by an interpreter on request.
Given the cluster RCT design, patient participants are not individually consented to randomisation; written informed consent for practices to participate in the trial is provided by the general practice lead acting as ‘guardian’ for patients in their care.
Written consent is obtained from patient participants for:
Participation in the MOPP questionnaire study (read and understood the participant information leaflet, voluntary participation, completion of baseline and 3,6,12-month follow up questionnaires)
Access to their electronic health record (EHR).
Written consent (optional) from patient participants is also requested for:
General practices are randomised (1:1) in balanced blocks, stratified by regional centre, by an independent statistician affiliated with Keele CTU. The randomisation sequence is generated using the randomisation lists module in the Power Analysis and Sample Size software (PASS: Sample Size Software | Power Analysis Software | PASS | NCSS.com). Allocation concealment is ensured by each practice having an anonymised code. Practices are randomised either to offer the PROMPPT review (intervention arm) or to continue with usual practice arrangements for reviewing patients prescribed opioids for persistent pain (control arm). Participants receive the care to which their practice is randomised.
Blinding for participating practice pharmacists and GPs is not possible. The main trial statistician, outcome data entry staff, and members of the research team involved in calculating daily morphine equivalent dose from self-reported pain medicines data are blinded to allocation (at cluster (practice) and individual participant level) until the analysis is finalised. The independent Trial Steering Committee (TSC) is also blinded to allocation, but unblinded data is shared with the independent Data Monitoring Committee (DMC) so that outcomes, such as patient safety, can be fully monitored.
The PROMPPT intervention, comprising a proactive practice pharmacist-led review for patients who have been prescribed opioids for at least 6 months and an associated pharmacist training package, is described below, informed by the TIDieR guidance29. Both the approach to delivering the PROMPPT review, and the content of the training package were shaped by earlier intervention development work17,19–22. They incorporate best practice guidance30,31, contemporary behaviour change theories and key communication skills to support shared decision-making32–35. The intervention aims to respect patient values and autonomy, ensuring that decisions about when and how to change are patient centred.
PROMPPT review. General practices in the intervention arm invite MOPP study participants, by letter, to arrange a review with the practice pharmacist. The invitation letter is accompanied by an information leaflet called ‘Getting Ready for Your Pain Review’. This leaflet aims to inform participants about what to expect at the initial review consultation and help them identify their priorities to discuss with the pharmacist during the consultation. The initial PROMPPT consultation (approximately 30 min) is conducted face-to-face or remotely (by video or telephone) according to patient preference.
The consultation begins with a holistic assessment of the participant’s pain and the impact of persistent pain on their life, followed by a personalised discussion to explore the participant’s personal experience of the effects (wanted/unwanted, useful/bothersome) of opioids. Pharmacists are encouraged to explore participants’ reasons for considering changing their use of opioids, their readiness to change and any ambivalence, before agreeing an individualised management plan.
The agreed plan may include opioid tapering, but this is not mandatory, for example if a participant obtains continued useful benefit from moderate opioid doses without experiencing troublesome side-effects. Where changes to medicines are agreed, goal setting is used to facilitate translation of intentions into action. Important barriers to reducing opioids specific to the individual, such as fear of pain worsening and/or withdrawal symptoms following opioid reduction, are addressed. Practice pharmacists are encouraged to signpost to information resources, which are hosted on a dedicated website. These resources include strategies for living well with pain and reducing opioids. Pharmacists may also signpost or refer to appropriate community services (for example physiotherapy and mental health support services) and, if needed for more complex cases, discussion/collaboration with the GP and/or referral to specialist services is advised. Follow-up is encouraged and arranged according to pharmacists’ clinical judgment and participant preference. Follow-up appointments, conducted face-to-face or remotely by video or telephone, according to pharmacists’ assessment of clinical need and patient preference, are anticipated to be shorter in duration (up to 15 minutes) on average. Pharmacists are also encouraged to agree and document a clear plan for contact, if needed, between scheduled appointments. A written summary of the agreed plan (‘Your Pain Action Plan’), including a link to the information resources webpage, is provided in either paper or electronic form, according to patient preference.
Practice pharmacists delivering PROMPPT are independent prescribers and have completed trial-specific training to deliver the PROMPPT intervention, as described below. Intervention fidelity is assessed in the process evaluation (described in detail below) using pharmacist completed intervention delivery templates and audio-recorded consultations.
Pharmacist training. A comprehensive training package, developed by experts in pain management, primary care clinicians, behaviour change experts and medical/pharmacy educationalists, aims to equip practice pharmacists to deliver PROMPPT reviews. The training comprises an e-learning course and two half-day online workshops delivered via Microsoft Teams. The e-learning course, delivered via the PROMPPT study website, includes video presentations by experts in pain management and behaviour change, as well as videos of four simulated consultations aligned with readiness to change35.
The training covers communication skills, how to conduct person-centred discussions about persistent pain and opioids, negotiating treatment plans, creating opioid tapering plans, optimising non-opioid pain management, supporting patients to live well with persistent pain, signposting to patient information resources and when to seek help (e.g. from a GP). Training also includes guidance to help practice pharmacists manage any emotional impact of conducting pain reviews.
The online workshops are facilitated by members of the research team with clinical and academic expertise in persistent pain management and behaviour change, supported by clinical champions, who are experienced clinical pharmacists and have completed the PROMPPT e-learning and received additional training in mentoring and supporting reflective practice. The online workshops incorporate role play, in simulated consultations, with feedback from the facilitators. In addition, practice pharmacists delivering PROMPPT reviews are mentored during the trial by one of the clinical champions.
Practice pharmacists are also provided with study-specific training including training on completion of study documentation, good clinical practice as applicable to research, and the maintenance of the study site file and study records. Reporting of serious adverse events and adverse events will also be covered.
MOPP study participants in control arm practices receive usual primary care arrangements for reviewing patients on long-term opioids for persistent pain, which are known to be variable10. Such reviews may be conducted by the GP or by another appropriately qualified healthcare professional and may take place as a face-to-face consultation or as a virtual review with the patient’s medication assessed from practice records without them being present.
Participating practices all have at least one practice pharmacist working in the practice at least some of the time. The roles adopted by practice pharmacists are variable, often including reviews for specific long-term conditions such as hypertension, diabetes and asthma, and Structured Medication Reviews (SMRs) for patients who may benefit from these, for example due to potentially problematic polypharmacy and / or use of potentially dependence forming medicines36. Since the PROMPPT trial was conceived, reducing high-risk opioid prescribing has been included in NHS England’s Medicines Safety Improvement Programme and, as a result, practice pharmacists may increasingly be conducting SMRs with patients who are prescribed opioids, but there is no specific training available regarding SMRs in this population and it remains unlikely that a practice pharmacist in a control practice would offer a structured medication review in the same way as a PROMPPT review without having received the PROMPPT training. Usual care arrangements are investigated as part of the process evaluation (described in detail below) to explore whether and how this affects outcomes.
There are three types of outcome data collection:
1. Individual participant data collected from baseline and follow-up (3, 6 and 12-month) questionnaires (online or postal).
2. Primary care electronic health record review.
3. Practice-level anonymised aggregated data of pain-related prescribing.
Baseline and follow-up questionnaires. The baseline and follow-up questionnaires are designed to collect information on descriptive characteristics of the participants and primary and secondary outcome measures (see Table 3). Patients are informed in their study invitation that they have been invited to take part in the Management of Opioids & Persistent Pain (MOPP) Study because they have long term pain and have been regularly prescribed opioid medicines.
| Assessments | Number of items | Timepoint | Minimum data collection for non- responders | |||
|---|---|---|---|---|---|---|
| Baseline | Follow-up | |||||
| 3-m | 6-m | 12-m | ||||
| Demographics | ||||||
| Date of birth | 1 | X | X | X | X | X |
| Sex | 1 | X | X | X | X | X |
| Ethnicity | 1 | X | ||||
| Socioeconomic status (Index of Multiple Deprivation (IMD)) | 1 | X | ||||
| Pain duration (years) | 1 | X | ||||
| Primary outcomes | ||||||
| Brief Pain Inventory37 (BPI) (total score) | 11 | X | X | X | X | X |
| Opioid use (mean daily morphine equivalent dose (MED)a | X | X | X | X | X | |
| Secondary outcomes | ||||||
| BPI37 pain severity | 4 | X | X | X | X | X |
| BPI37 pain Interference | 7 | X | X | X | X | X |
| Non-opioid pain medicines use (past week)b | X | X | X | X | X | |
| Opioid-related side-effects (Checklistc) | 10 | X | X | X | X | |
| Depression (Patient Health Questionnaire (PHQ-8)38) | 8 | X | X | X | X | |
| Anxiety (Generalised Anxiety Disorder Questionnaire (GAD-7)39) | 7 | X | X | X | X | |
| Confidence to cope with pain (Pain Self- Efficacy Questionnaire40) | 10 | X | X | X | X | |
| Health-related quality of life (EQ-5D-5L)41 | 5 | X | X | X | X | X |
| Work (work status, occupation, time off work, presenteeism)42,43 | 5 | X | X | X | X | |
| Healthcare resource use (NHS/private & out of pocket expenses)d | 7 | X | X | X | ||
a Daily MED calculated from self-reported pain medicines use questionnaire, with any missing information regarding drug name/strength obtained from the electronic health record (EHR).
b Obtained from self-reported pain medicines use questionnaire, with any missing data on prescribed non-opioid pain medicines obtained from EHR.
c Checklist of opioid-related side-effects (presence & severity) developed from existing measures44–46
d Healthcare resource use questionnaire developed for this research and refined following a feasibility study22
To maximise data collection at all follow-up timepoints, non-responders (online) receive a reminder by email (with text alert) after 1 week and a postal questionnaire after 2 weeks. Non-responders (postal) receive a reminder postcard after 2 weeks and a new paper questionnaire after 4 weeks. All non-responders to the reminders are contacted by telephone by a blinded trial administrator for minimum data collection (MDC) after 6 weeks. A brief MDC questionnaire is sent by post to those who cannot be contacted after 3 telephone attempts (Amendment 4, Table 1). MDC collects primary and key secondary outcome data (opioid and non-opioid pain medicines use, Brief Pain Inventory (BPI) and EQ-5D-5L) along with date of birth and sex to ensure the data are provided by the intended participant. Participants opting to complete the questionnaire online are advised, in the participant information leaflet, that they can stop completing the questionnaire at any time point, without giving a reason, but that answers entered up to that point are retained unless they contact the study team to opt out of this (Amendment 4, Table 1). Where there are ambiguous or incomplete questionnaire entries that interfere with calculation of daily opioid use, a member of Keele CTU staff attempts to contact participants by telephone or in writing to obtain the missing data. Incomplete or ambiguous questionnaire entries regarding prescribed pain medicines can also be checked with the electronic health record, to obtain the missing data e.g. regarding drug strength, if necessary.
Medical Record Review (MRR). With participant consent, data is extracted from electronic practice health records to capture data on their prescriptions from baseline (defined as the period 90 days up to the date of their recruitment) up to the date of their 12-month follow-up to facilitate checking of ambiguous or incomplete questionnaire entries regarding pain medicines use if needed. MRR also captures information about PROMPPT-related consultations.
Each participating practice provides anonymised electronic prescribing data for registered adult patients, aged ≥18 years, who are not receiving palliative care for cancer (defined as patients without both a code for cancer and for palliative care). We will compare prescribing of opioids and non-opioid medicines commonly prescribed for patients with persistent pain (specifically paracetamol, topical pain treatments, non-steroidal anti-inflammatory drugs (NSAIDs), nefopam, gabapentinoids (gabapentin, pregabalin), antidepressants, benzodiazepines and Z-drug hypnotics (zopiclone, zolpidem) at baseline (defined as the period 90 days up to the date the first participant was recruited at that practice) and 12-month follow-up (defined as the period 90 days up to the date 12 months after the first participant was recruited at that practice).
When evaluating interventions aimed at reducing opioid use it is recommended to also measure the impact on pain47, therefore we chose two co-primary outcomes, measured at 12-month follow-up:
1. Reduction in opioid use (participant achieves at least a 25% reduction in daily morphine equivalent dose (MED) from their baseline (binary outcome, yes or no))
AND
2. Non-inferiority of the Brief Pain Inventory (BPI)37 total score
Members of the research team calculate a daily MED from self-reported pain medicines use data in participant questionnaires using a calculator created in Microsoft Excel (2016) by lead author JA. The calculator uses published opioid conversion factors48–50 (see Table 4) and was refined following testing with members of the research team (SAH, TH, SW, RK) in a feasibility study22. As there is currently no accepted definition of a clinically meaningful reduction in opioid use47,51, it was agreed with the study funder that, based on clinical experience, a 25% reduction in opioid use from their baseline reflects a clinically reasonable minimum threshold across a range of opioids and doses.
| Conversion factor* | Equivalent dose to 10mg morphine | |
|---|---|---|
| Oral preparations (mg/day) | ||
| Codeine | 0.1 | 100mg |
| Dihydrocodeine | 0.1 | 100mg |
| Dextropropoxyphene | 0.23† | 43.5mg |
| Tramadol | 0.1 | 100mg |
| Tapentadol | 0.4 | 25mg |
| Oxycodone | 1.5 | 6.6mg |
| Pethidine (Merperidine) | 0.1† | 100mg |
| Hydromorphone Methadone | 5.0 3.0† | 2mg |
| Sublingual preparations (mg/day) | ||
| Buprenorphine | 40‡ | 0.25mg |
| Transdermal patches (micrograms / hour) | Morphine equivalent (mg/day) | |
| Buprenorphine (5) | 2.4 | 12 |
| Buprenorphine (10) | 2.4 | 24 |
| Buprenorphine (20) | 2.4 | 48 |
| Fentanyl (12) | 2.5 | 30 |
| Fentanyl (25) | 2.4 | 60 |
| Fentanyl (50) | 2.4 | 120 |
| Fentanyl (75) | 2.4 | 180 |
| Fentanyl (100) | 2.4 | 240 |
*Conversion factors are approximations of equianalgesic doses and there is some variation across different sources. The conversions factors used in this study are those accepted by the UK Faculty of Pain Medicine, Royal College of Anaesthetists (Opioid Aware49), except for dextropropoxyphene, pethidine, methadone and sublingual buprenorphine, which are not included in the Opioid Aware list. The source of conversion factors for these drugs are below
†CONSORT morphine equivalent conversion factors48 (dextropropoxyphene, pethidine, methadone)
‡Faculty of Pain Medicine, Australian and New Zealand College of Anaesthetists (ANZCA)50 (sublingual buprenorphine)
The BPI is a composite measure of pain intensity and pain-related functional interference, recommended for use in chronic pain trials and validated in primary care populations with persistent pain52,53. The total score reflects the multidimensional impact of pain and has comparable responsiveness to the BPI pain intensity and interference subscales in this population54,55. In defining the non-inferiority margin for the BPI total score we considered the following: (1) Feedback from patients that pain and pain-related interference does not noticeably increase as a result of reducing opioids and, (2) the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT)52,53 international consensus recommendations on interpreting the clinical importance of treatment outcomes in chronic pain trials. IMMPACT identified a 1-point change on the BPI pain intensity (0-10) scale as the smallest noticeable change in pain and a 1-point change on the BPI pain interference (0-10) scale as a clinically meaningful change53. Therefore, our non-inferiority margin for the BPI total score needed to be less than 1.0. We chose 0.6 as a difference that is considered clinically non-inferior and aligns with the standard error of measurement of the BPI total score54.
Our non-inferiority margin translates to an effect size of 0.3 (0.6/2) if a standard deviation for the BPI of 253 is assumed. We chose not to lower the non-inferiority margin further, for example to give a small effect size of 0.2 (based on effect size criteria from Cohen56), as recent IMMPACT guidance on evaluating opioid reduction in clinical trials suggests that a somewhat larger non-inferiority margin can be considered for pain in ‘opioid sparing’ trials because of the potentially large benefits of reducing opioid use (e.g. reducing opioid-related adverse effects)47.
To facilitate comparison and pooling of our results, our secondary outcomes are informed by IMMPACT recommendations for trials of interventions for persistent pain and opioid sparing47,52,53. These include:
Pain severity, assessed using the BPI37 pain severity subscale.
Pain-related interference, assessed using the BPI37 interference subscale.
Daily opioid use (mean daily MED calculated as described above).
Non-opioid pain-related medicines use (yes or no) of paracetamol, topical pain treatments, NSAIDs, nefopam, gabapentinoids, antidepressants, benzodiazepines and z-drug hypnotics (zopiclone and zolpidem), assessed from the self-reported pain medicines questionnaire.
Opioid-related side-effects (presence and severity) assessed from a self-reported side-effects checklist, developed for this research programme from existing measures44–46.
Depressive symptoms assessed using the Patient Health Questionnaire Depression Scale (PHQ-8)38, a validated 8-item questionnaire to assess depressive symptomology over the past 2 weeks.
Anxiety symptoms assessed using the Generalised Anxiety Assessment (GAD-7)39, a validated 7-item questionnaire.
Confidence to cope with pain (pain self-efficacy), assessed using the validated 10-item Pain Self-Efficacy Questionnaire (PSEQ)40.
Health-related quality of life assessed using the EQ-5D-5L41.
All outcome measures and data collection timepoints are presented in Table 3.
Questions on additional persistent pain-related NHS and private practice health care appointments are included in the 3, 6 and 12-month follow-up questionnaires. The health care resource use questionnaire was developed for this research programme, tested in the feasibility study22 and refined considering the findings. Resource use information is obtained on primary care visits, (e.g. general practitioners, practice nurses, and physiotherapy), visits to other health care professionals (e.g. osteopath, chiropractor, psychologist / counsellor), pain medicines (prescribed and non-prescribed) including opioids, non-opioid analgesics and drugs to manage opioid-related side-effects e.g. laxatives and antihistamines, tests and investigations, non-drug treatments (e.g. acupuncture, injections), secondary care consultations, inpatient stays, and surgery during the previous 6 months. Given the nature of persistent pain, broader costs related to both out of pocket costs (e.g. over the counter medicines such as laxatives) and productivity losses (e.g. time-off work related to pain and reduced work performance (presenteeism) are also included. Information on time off work, occupation, typical work activities and the nature of their employment (full time or part time) is requested, and the Single-Item Presenteeism Question from the Work Productivity and Activity Impairment Questionnaire42,43 is used to estimate productivity losses relating to presenteeism. The average wage for each respondent will be identified using UK Standard Occupational Classification coding and annual earnings data for each job type. The outcome of interest for the economic analysis is the quality-adjusted life years (QALYs) and these will be calculated using EQ-5D-5L41 responses obtained at baseline, 3, 6, and 12-months follow-up.
The clinical management recommendations given to participating practice pharmacists and GPs in intervention arm practices represent evidence-based best practice and have strong clinical community endorsement and credibility. Therefore, the potential harms of this study are minimal.
A Serious Adverse Event (SAE) is defined by the Health Research Authority (HRA) as an untoward occurrence that: results in death, is life-threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, consists of a congenital anomaly or birth defect; or is otherwise considered medically significant by the investigator. All SAEs, either confirmed or suspected, are communicated to Keele CTU within 24 hours. The Chief Investigator (CI) or Associated Investigator (AI) is responsible for conducting a causality assessment. Suspected or confirmed SAEs are reviewed by the Data Monitoring Committee and reported to the Trial Steering Committee. SAE’s that are assessed as related to the trial and unexpected are reported to the Research Ethics Committee.
Our first co-primary outcome is opioid (daily MED) reduction, by at least a 25% (yes/no), between baseline and 12 months. We judged from our clinical experience that a 20% difference between the intervention and control arms in the proportion of patients reporting ≥25% reduction in opioid use represents a meaningful difference. Therefore, assuming 40% of the control arm and 60% of the intervention arm reduce opioid use according to that definition over 12 months, we estimated that a total sample size of 260 would be needed for a non-cluster RCT (power 90%, two-tailed statistical significance 5%). Our assumption that 40% in the control arm would reduce opioids was based on our previous (unpublished) analysis of opioid prescribing data from the Clinical Practice Research Datalink (CPRD) indicating that approximately 20% of patients who have been prescribed opioids for 6 months or longer stop taking opioids completely without any intervention and our assumption, based on clinical experience, that a further 20% of the control arm, whilst not stopping completely, would reduce their opioid use according to our definition.
For the second co-primary outcome (total BPI), as explained above, we assumed a 0.6-point non-inferiority margin and a standard deviation of 2. We estimated the standard deviation from published literature54,57 and from our feasibility study22. With power 90%, one-tailed statistical significance 2.5%, a sample size of 468 would be needed for a non-cluster RCT.
Given the co-primary study hypotheses, we considered that power for our study may not be persevered at 90%. However, given that sample size differs considerably between the two hypotheses the impact on study power is lessened. Conservatively assuming that the two co-primary outcomes are not correlated, using the intersection-union test58,59, the overall power for the study is 0.89. As 0.89 is close to 0.9, the sample size was not adjusted to account for the loss of power from testing a co-primary hypothesis.
Given the cluster design, our sample size was inflated to account for clustering. Assuming (i) an intra-cluster correlation coefficient 0.01 (estimated from our POST trial and other similar trials in primary care60–62), (ii) a coefficient of variation 0.40 (estimated to be slightly lower than other estimates in primary care trials63 to reflect that our study design caps the number of patients invited per practice), and (iii) an average of 30 patients recruited per practice (estimated from our feasibility study22), this translated to a design effect of 1.3464. Given a 20% loss to follow-up at 3 months in our feasibility study, we conservatively assumed a 30% loss to questionnaire follow-up at the later 12-month follow-up and therefore the sample size for the individually randomised RCT was inflated by 1.91 and we planned to recruit 896 patients (448 per arm).
Based on our feasibility study22, we estimated that, on average, 2.5% of patients on a practice list would be eligible for our study, and that from a practice with 8000 patients, 200 would be invited to participate in the study and 15% (n=30) would consent and return a baseline questionnaire. Therefore, we initially planned to recruit 30 general practices, with a plan to review our recruitment assumptions following the internal pilot study. The 3-month follow-up rate in the internal pilot was slightly lower than the 70% we had assumed at 12 months. To mitigate this, we increased the number of GP practices included in the study (from 30 to 38) to maintain the desired level of power for the study at 90%.
A priori data analysis plans will be agreed with the independent TSC and DMC prior to database lock and will form the definitive analysis plan for the trial. An outline of the planned statistical analysis is given below:
A CONSORT flow diagram will be produced to document the flow of clusters and participants through the trial and will include reasons for withdrawal if given. Descriptive statistics will be used to describe key patient and practice-level baseline variables overall, by treatment arm (to explore balance of characteristics across randomised treatment arms) and by whether participants returned a 12-month follow-up questionnaire (to explore the impact of loss to follow-up).
Primary analysis will use multilevel linear (BPI) and logistic (opioid reduction) regression to account for the clustering in the data and will compare the effectiveness of providing the PROMPPT intervention with usual primary care arrangements for review of patients prescribed opioids for persistent pain at 12 months, adjusting for baseline BPI score (for the BPI outcome) and baseline daily MED (for the opioid reduction outcome). The superiority and non-inferiority hypotheses will be assessed simultaneously; superiority will be concluded if the 95% two-sided confidence interval for the opioid reduction treatment effect does not contain one and non-inferiority concluded if the upper limit of the 95% two-sided confidence interval for the mean difference in BPI score is <0.6.
The primary analysis will be conducted using data from all clusters and patients randomised into the trial. A secondary analysis will be conducted to compare the effectiveness of accessing the PROMPPT intervention compared to usual primary care in participants who attended a PROMPPT review.
Secondary (longitudinal) multilevel analyses, based on self-report questionnaire data, will explore whether change in the BPI total score (the co-primary outcome measure) and the secondary outcomes (BPI pain, BPI pain interference, daily MED, non-opioid pain medicine use, depression, anxiety, pain self-efficacy and treatment side effects) differ by treatment arm over time (i.e. using data from all time-points). The models will include a term to account for clustering by general practice and a time by treatment interaction. The beta coefficient and 95% confidence interval for the interaction term will be reported and graphical methods used to display the nature of any interactions found in the data. The continuous outcome, percentage change in daily MED (baseline to 12-month follow-up), will also be analysed using multilevel linear models, but as the data will be a single observation per participant, this model will not include a term for “time” in the multilevel model.
Descriptive statistics and 95% confidence intervals will be used to describe the aggregate data from the pseudonymised electronic health records at baseline and 12-months, to include a comparison of opioid and non-opioid analgesic use by treatment arm and to explore within-practice changes in between baseline (defined as the period 90 days up to the date the first participant was recruited at that practice) and 12-month follow-up (defined as the period 90 days up to the date 12 months after the first participant was recruited at that practice).
Subgroup analyses will explore whether the conclusions of the primary analysis for opioid reduction depends on baseline levels of pain self-efficacy and baseline daily MED. This will be explored by including an interaction term in the primary model between treatment and the subgroup covariate of interest, and by running separate models for each covariate.
The primary analysis will be adjusted for key a priori covariates specified in the analysis plan but are likely to include (1) stratification factors included in the randomisation process and (2) key patient-level variables likely to impact on outcome e.g. deprivation, age and sex.
Missing data will be handled by using multi-level models as the analysis method or by multiple imputation of missing data as appropriate.
An incremental cost-utility analysis will be undertaken from a base-case NHS/Personal Social Services (PSS) perspective to estimate the cost per quality adjusted life year (QALY) over 12 months follow-up, using patient level data on costs and trial outcomes. Alternative perspectives will include a health care and societal perspectives, taking into account private expenditures and out of pocket cost and productivity losses due to time off work.
The outcome of interest for the economic analysis is quality-adjusted life years (QALYs) and these will be calculated using EQ-5D-5L responses obtained in at baseline, 3, 6, and 12-months follow-up.
In line with updated NICE recommendations, individual EQ-5D-5L responses will be converted into index scores using a mapping algorithm developed by the Decision Support Unit. The 'EEPRU dataset' will be employed to convert EQ-5D-5L questionnaire responses onto the EQ-5D-3L utility scores65.
Cost data is likely to have a skewed distribution and therefore a non-parametric comparison of means (e.g., bootstrapping) will be undertaken to estimate 95% confidence intervals around costs. Multilevel modelling statistical techniques, taking into consideration clustering and correlation in the cost and effect data will be considered in line with standard literature. QALYs will be calculated from EQ-5D-5L responses, using the “area under the curve” approach. Unit costs will be applied to all health care resource use items, and mean resource use (for each health care use category) and mean total costs will be calculated for all trial participants. Analysis of productivity losses will use the human capital approach. Multiple imputation will be used to impute all missing values for the EQ-5D-5L and total cost estimates for non-responders to ensure all participants are included in the analysis.
A cost-consequence analysis will initially be reported, describing all the important results relating to costs and consequences (across the full range of clinical and cost outcomes). Subsequently, incremental cost-utility analysis will be undertaken to estimate the incremental cost per QALY gained, adjusting for baseline covariates in line with the primary clinical evaluation. The analysis will be conducted on an intention to treat basis, and in accordance with current cRCT guidelines66. This analysis will involve adopting multilevel modelling statistical techniques taking into consideration clustering in the cost and effect data67. Separate generalised equation models, controlling for clustering will be used to estimate the mean incremental costs and QALYs for the intervention relative to the control.
The robustness of the results will be explored using sensitivity analysis. This will explore uncertainties in the methods employed to analyse the data (for example, a complete-case analysis as an alternative to using a multiple-imputed data set, and in estimation of costs), and any assumptions made in the analysis. Cost-effectiveness acceptability curves will be generated to reflect the probability the intervention will be cost effective at different cost per QALY willingness to pay thresholds. If the intervention is shown to be clinically and cost-effective within the trial, a simple budget impact analysis will be undertaken to calculate the potential overall cost for a typical primary care practice, in terms of delivering the intervention and potential cost savings from reduced opioid prescribing.
Decision modelling, using a Markov model will also be undertaken to estimate the potential longer-term cost-effectiveness of the intervention. The model-based analysis will be performed from an NHS perspective with appropriate discounting of costs and outcomes at 3.5%. The model will assess the impact of the intervention on quality of life, continued opioid use, escalation of opioid medication, and the potential for opioid-related serious adverse events. The model will be populated using the trial data, literature review of previously published relevant models, routine datasets and where appropriate consultation with clinical experts and Patient and Public Involvement (PPI). The model will be subject to deterministic sensitivity analysis by changing individual parameter values, broadening the perspective and changing model assumptions. Model parameters will be inputted as distributions and probabilistic sensitivity analysis will be undertaken to simultaneously incorporate all parameter uncertainty. Cost-effectiveness planes and cost-effectiveness acceptability curves will be presented to show the probability the intervention is cost-effective at different cost/QALY thresholds. Full details of the decision model methods, time horizon, structure and parameters will be detailed in the final health economics publication.
To provide greater confidence in conclusions about effectiveness of the PROMPPT intervention and the generalisability of the results, a process evaluation using a mixed methods approach68 is being conducted. In line with MRC guidance on the evaluation of complex interventions69, the objectives, aligned to the key domains of implementation, mechanisms of impact and contextual factors, are:
1. To assess how PROMPPT is implemented in and adopted by clusters and assess the processes by which practice pharmacists and GPs integrate and adopt PROMPPT related work into existing systems and everyday work.
2. To investigate intervention fidelity by observing what is delivered in each intervention cluster and what practice pharmacists do because of PROMPPT. This will highlight how closely the delivery of PROMPPT matches what was intended.
3. To investigate how patients with persistent pain respond to and engage with PROMPPT and what mechanisms of impact lead to change.
Additionally, given the rapidly changing landscape in primary care, in particular relating to the role of practice pharmacists in medicines optimisation, we recognised the importance of examining current usual care regarding review of patients prescribed opioids for persistent pain and the evolving role of practice pharmacists with clinicians from clusters in both trial arms, and topic guides evolved accordingly.
Patient participants. Participants from intervention arm clusters who provide written consent to being contacted about a future related research study and subsequently attend a PROMPPT review are invited to participate in the MOPP-2 study. Participants are invited to complete an acceptability questionnaire (described below) and/or take part in an interview (up to n=20) and may choose to consent to any, all or none of these options. Additionally, all participants who do not take up the PROMPPT review are invited to participate in an interview to explore their reasons for declining. Written consent is obtained from all participants in the process evaluation.
Clinicians (GPs and practice pharmacists). All practice pharmacists from intervention arm practices are asked to consent to audio-recording of a sample of PROMPPT consultations and to an interview. A sample of GPs from intervention arm practices (up to n=15) and practice pharmacists and GPs from control arm practices (up to n=12) are also invited to take part in an interview.
PROMPPT clinical champions. All PROMPPT Clinical Champions are interviewed to explore their experiences of mentoring practice pharmacists during the delivery of PROMPPT.
Audio-recorded consultations. A sample of approximately 10% of PROMPPT consultations are digitally audio-recorded to allow assessment of key aspects of intervention delivery70: treatment differentiation (Did the practice pharmacists only deliver PROMPPT and not other treatments?), treatment competency (Did practice pharmacists maintain the skills learned in training?), and treatment delivery (Were PROMPPT components delivered as intended?).
The research team work with participating practices to identify and approach suitable participants who have scheduled a PROMPPT consultation. Informed, written consent to audio-record the consultation is obtained from patient participants by a researcher prior to the consultation and confirmed with practice pharmacists prior to the start of the consultation. This process was used successfully in the feasibility study22. Audio-recording files are securely sent to Keele CTU for analysis.
Case-report forms. Practice pharmacists in the intervention clusters are required to complete an electronic case report form (eCRF) following each PROMPPT consultation. The eCRF collects information on what happened during the PROMPPT consultation (e.g. discussing the patient’s pain story and the impact of their pain, discussion of the patient perspective on making changes to opioid medicine, developing a personalised pain action plan) and will be used to assess intervention fidelity (i.e. Was PROMPPT delivered as intended?) and dose (How much was delivered?).
Semi-structured interviews. Consenting participants in the intervention arm are interviewed after attending at least one PROMPPT consultation. The interviews are based on semi-structured topic guides developed with our PPI group and include questions informed by our programme theories regarding behaviour change (Theoretical Domains Framework (TDF))71, implementation (Normalisation Process Theory (NTP))72,73 and acceptability (Theoretical Framework of Acceptability (TFA))74. Consenting participants who decline a PROMPPT review, and accept an invitation to speak to a researcher, are interviewed using a semi-structured topic guide that will explore reasons for declining. A similar process is followed for the clinicians, whereby they are interviewed once they have experience of PROMPPT intervention delivery. Topic guides are refined, as needed, during data collection to allow any emerging issues to be explored.
The sample sizes outlined above are expected to be sufficient to achieve information power75, providing appropriate range and depth of insights, whilst being achievable within the scope of the project. Similar topics are covered with all interviewees to ensure comparisons between their responses, while at the same time enabling them to reflect on their specific expectations, understandings and experiences.
Informed consent is obtained prior to all interviews commencing. All interviews are conducted remotely via telephone by an experienced qualitative researcher, and audio-recorded with participants’ consent. The interviews are securely sent to a professional transcription company to be transcribed verbatim. Transcripts are checked for accuracy and anonymised by members of the research team ahead of analysis.
Acceptability questionnaire. All participants who attended the initial PROMPPT review are sent a short survey (known as the Acceptability Questionnaire) about their experiences of the consultation. The Acceptability Questionnaire comprises 12 questions from two existing measures: a modified version of the treatment acceptability and credibility measure76 and the Theoretical Framework of Acceptability (TFA) questionnaire74.
The modified acceptability and credibility measure, which has been used in previous similar studies77,78, includes 4 items (scored on a 0-10 scale) and will assess (1) how logical the PROMPPT review seems to participants, (2) how confident participants are that it will be successful in helping them, (3) how confident participants would be in recommending it to a friend, and (4) how satisfied they were with the intervention overall.
The TFA questionnaire74 comprises 8 items (scored on a 0-5 scale). The first is a global acceptability question and the remainder represent key constructs of acceptability relating to healthcare interventions (affective attitude, burden, intervention coherence, ethicality, perceived effectiveness, opportunity costs, and self-efficacy).
Practice profile form. A practice profile form is completed by all participating GP practices at multiple time points across the trial, including at the beginning and towards the end. It collects information about current practice activity relating to persistent pain management, practice pharmacist workload and responsibilities within the practice, and practice size and organisational characteristics. These data will facilitate assessment of practice context and effects of any variation in context.
Audio-recorded consultations. Researchers from the process evaluation team will examine the audio-recordings from the PROMPPT consultations and use a previously developed tick box score sheet to assess whether components of the consultation intended to be included, and focused on during training, were demonstrated by the practice pharmacists.
eCRFs. The intervention delivery template will be reviewed for each participant and the proportion of times that each component of the intervention was used will be reported. A judgement will also be made as to whether the patient received the intervention as intended, in line with the training, and whether the patient attended all scheduled follow-up appointments. The proportion of patients being treated in line with the training will then be calculated out of all participants who attended the first appointment with the practice pharmacist. For audio-recorded consultations, corresponding eCRFs will be triangulated with the audio-recordings to assess fidelity of the training and intervention.
Semi-structured interviews. The Framework approach79 to analysis will be used starting deductively from pre-set aims and objectives, will enable a focused and efficient analysis within the timeframe of this study80, and can be used within a multidisciplinary research team setting79. Analysis will involve familiarisation, identification of an analytical framework (e.g. study aims, TDF71, TFA74 and NPT72,73,81), indexing, charting and mapping, and interpretation82. Analysis will include members of the research team from different professional backgrounds (e.g. GPs and pharmacists) to increase trustworthiness83. Continuous team analysis of data will help to challenge interpretations, and coding will be refined and agreed through ongoing discussion84. The data analysis will be facilitated using qualitative data-analysis software (NVivo: Qualitative Data Analysis Software Version 1485.
Acceptability questionnaire. The mean and standard deviation, as well as the median and interquartile range, for the TFA questions and the acceptability/credibility questions will be calculated from items 1-8 and items 9-12 of the Acceptability Questionnaire respectively. A mean score of ≥ 5/10 for each of items 9-12 will be considered the threshold for acceptability/credibility, as in previous trials using this measure77,78.
Practice profile form. A narrative review of practice profile form responses from participating practices will be undertaken. Responses from multiple time points across the trial will be examined and descriptive summaries produced to facilitate assessment of practice context and effects of variation in context.
Integrated analysis. When analysis of the quantitative and qualitative data is complete, a triangulation protocol68,86 will be used. This technique enables integration of data in order to investigate completeness, convergence, and dissonance of key themes across datasets87. Methods include following a thread and development of a convergence coding matrix. The matrix allows findings from different study components to be displayed side by side. Integration will aid interpretation of findings on implementation, acceptability, fidelity, mechanisms of impact and context.
The PROMPPT Trial Management Group (TMG), comprising members of the research team and Keele CTU, has overall responsibility for the set-up, promotion, ongoing management and monitoring of the trial, and for the analysis and interpretation of results. The TMG meets regularly to monitor protocol compliance of recruitment, intervention delivery and follow-up procedures; data collection and database development; completion of regulatory and funder reporting requirements and appropriate reporting of adverse events. All data collection, database design, data input and cleaning, as well as trial oversight procedures, are managed in accordance with Keele University’s Health and Social Care Research (HSCR) Quality Management System (QMS) and Keele CTU’s QMS. An independent Trial Steering Committee (TSC) was established to provide independent oversight of the trial. The TSC is chaired by a Professor of Primary Care and includes a statistician, a pharmacist, and a patient representative. The TSC was consulted during protocol development and met to approve the final protocol prior to submission for ethical approval, and at agreed intervals over the course of the trial. An independent Data Monitoring Committee (DMC), comprising a statistician and academics with clinical expertise in pharmacy and general practice, was also convened and is responsible for scrutinising trial data, with oversight responsibility for the safety of participants. Detailed reports, focusing on recruitment, retention, protocol compliance, protocol amendments and adverse events are prepared for the TSC and DMC by Keele CTU and members of the research team.
All data collection, database design, data input and cleaning, as well as trial oversight procedures, are in line with Keele University’s HSCR QMS and Keele CTU QMS and the conditions of the funding body. Data will be centrally monitored for quality and completeness by Keele CTU.
All data collected during the trial are kept strictly confidential and are handled and stored in line with Keele University and Keele CTU Data Security procedures, Keele University’s HSCR QMS and Keele CTU QMS, which are in accordance with the Data Protection Act (2018) and UK General Data Protection Regulation (GDPR). If a participant withdraws consent for further data collection, their data up to that point remains on file and will be included in the final analysis.
At the end of the study, data will be securely archived in line with Keele University HSCR and Keele CTU Standard Operating Procedures (SOPs) for 10 years after submission of the End of Study Declaration. Data held by Keele CTU will be archived in the designated Keele CTU archive facility and site data and documents will be archived at the participating sites. Following a retention review, if the archived material is agreed to be destroyed, arrangements for confidential destruction will then be made.
Any requests for access to the data from anyone outside of the research team (e.g. collaboration, joint publication, and data sharing requests from publishers) will follow the Keele CTU SOP on data sharing.
Recruitment of general practices commenced on 20 May 2022 and participant recruitment started on 21 June 2022.
An internal pilot study analysed recruitment, intervention uptake and fidelity of intervention delivery against pre-specified progression criteria between 20 May 2022 and 22 December 2022 from participants in the first batch of GP practices recruited and assessed retention at 3-month follow-up. Recruitment exceeded the pre-specified targets with 14 practices (target 12) and 388 participants (target 350, 50 per month on average) recruited. Fidelity of intervention (PROMPPT review) delivery was good with 89% of pain reviews delivered in line with the training (target 80%) based on the intervention delivery template. Intervention uptake was slightly lower at 62% than the pre-specified target of 70%. However, the first practice to invite participants sent all the invitations at once and had particularly low uptake (32%) which lowered the overall rate. Subsequently, practices were advised to send the review invitations in batches, according to pharmacist availability, which made it easier for practices to monitor uptake and send reminders. Rates of retention at 3-month follow-up were lower than expected, with 106 out of 174 (61%) of participants with sufficient time (8 weeks) to respond returning the 3-month follow-up questionnaire, whilst the sample size estimate assumed 70% retention at 12-month follow-up. These findings were reported to the DMC and TSC (March 2023). Both were satisfied that the trial should continue, and a range of measures were agreed and implemented to improve retention at follow-up and maximise primary outcome data collection at 12 months (See Amendment 4. Table 1). To mitigate any impact of lower follow-up rates on study power we also increased the number of general practices in the trial from 30 to 38 (Amendment 7. Table 1).
Dissemination of the results of the PROMPPT trial will be supported by Keele University’s Impact Acceleration Unit. The findings will be made widely and freely available to all stakeholders in ways that are easy to access at no cost. Our PPI Group will advise on how to translate these into easily understandable messages and on how best to disseminate the results to the wider public. In addition to publications in open-access peer-reviewed journals and conference presentations, we will use our website, NHS networks, links to professional bodies and social media to support dissemination of the findings to all stakeholders.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)