Keywords
Vagus nerve stimulation, fatigue, post-COVID, dysautonomia, statistical analysis
Vagus nerve stimulation, fatigue, post-COVID, dysautonomia, statistical analysis
Post-COVID fatigue (pCF) is one of the most common and persistent complications of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, whether severe, requiring hospital admission, or mild, and has become a major public health concern. Estimates from the Office for National Statistics indicate that around 2.3% of the UK population experience ongoing fatigue after COVID-19 infection.1 People with pCF often report profound physical tiredness as well as cognitive and mental fatigue, including difficulties with concentration, memory, and sustained effort, leading to substantial impairment in daily functioning and quality of life.2,3
Current management strategies for pCF have largely been adapted from approaches used in chronic fatigue syndrome (CFS), such as graded exercise therapy and cognitive behavioural therapy. These approaches are based on the assumption that post-viral fatigue represents a subacute form of CFS. However, there is currently little evidence to support their effectiveness specifically in people with post-COVID fatigue, and patient feedback frequently highlights persistent symptoms and unmet clinical need.2,4
Recent mechanistic work has provided evidence that fatigue in pCF is associated with abnormalities across multiple components of the nervous system. Baker et al.5 demonstrated that individuals with pCF show dysregulation within the central, peripheral, and autonomic nervous systems, with abnormalities observed across a range of physiological, neurophysiological, and behavioural measures. These findings suggest that fatigue in pCF has a biological basis involving disrupted neuromodulatory and autonomic pathways.
The vagus nerve is a major component of the autonomic nervous system and plays a key role in metabolic homeostasis and immune regulation through the cholinergic anti-inflammatory reflex.6,7 Stimulation of the vagus nerve is known to influence central adrenergic, serotonergic, and cholinergic pathways,8,9 and vagal dysfunction has been proposed as a potential contributor to fatigue in post-COVID states. Vagus neuropathy has also been reported following COVID-19 infection and may be associated with more severe disease.10,11
Vagus nerve stimulation (VNS) is a well-established therapy in other clinical conditions, such as refractory epilepsy, but conventional VNS requires surgical implantation of electrodes and is therefore not suitable for widespread use in people with pCF. More recently, transcutaneous auricular vagus nerve stimulation (taVNS) has emerged as a non-invasive alternative that targets the auricular branch of the vagus nerve. This approach can be delivered using a standard transcutaneous electrical nerve stimulation (TENS) device, allowing safe self-administration at home. Evidence from autoimmune conditions associated with fatigue, including primary Sjögren’s syndrome and systemic lupus erythematosus, suggests that non-invasive vagus nerve stimulation may lead to improvements in fatigue symptoms.12 However, robust evidence for its effectiveness in post-COVID fatigue is currently lacking.
The PAuSing-pCF trial13,14 was designed to address this gap in the evidence. This single-site, single-blind, randomised, sham-controlled study evaluated whether transcutaneous auricular vagus nerve stimulation, self-administered using a TENS device, could reduce symptoms of fatigue in adults with post-COVID fatigue. In addition to assessing changes in fatigue severity using frequent Fatigue Visual Analogue Scale measurements, the study also explored whether neurophysiological measurements predicted response to stimulation and examined the optimal duration and dose of non-invasive vagus nerve stimulation. In this article, we report the between-group differences within the PAuSing-pCF trial and assess both the clinical and mechanistic effects of taVNS in post-COVID fatigue.15,16
PPIE (patient and public involvement and engagement) was critical to the concept, design and delivery of the research. Patients with post-COVID fatigue (pCF) were first engaged in preliminary discussions around the concept and design of the study in 2020–2021, as participants in a separate research project at Newcastle University, funded by the MRC, to investigate neural mechanisms of pCF (MR/W004798/1), before the PAuSing-pCF study had been conceived. The consensus feedback from these patients confirmed the priority placed on identifying treatments for pCF and a preference for non-pharmacological treatments.
In the first month of the project, PPIE meetings were convened. These were run remotely, over Zoom or MS Teams platforms (Newcastle University institutional licences), which because of the COVID-19 pandemic had become both more widely available and a more familiar medium for communicating/meeting. These platforms made PPIE meetings much easier to arrange and were the preferred option for patients/stakeholders.
PPIE meetings comprised focus groups aimed at refining practical aspects relating to the delivery of the study, and specific to individuals experiencing disabling post-COVID fatigue (pCF); for example, the timing of assessments (morning or afternoon). Focus groups were also asked to consider and express preferences with regards to the study design; for example, patients were unanimous in their support for an open label phase in the interventional study design.
Before running focus groups, an outline of the study design was sent to participants and a link to an online survey comprising specific questions pertaining to the study and some free text comment boxes. This survey, designed by the study PPIE panel convened specifically for this purpose, informed discussions in the PPIE focus groups. The study PPIE panel also discussed/co-designed subsequent PPIE surveys sent to patient groups.
Feedback from the PPIE survey and focus groups was critical to the design and delivery of the study, particularly in determining the optimum number of assessments at each study visit, which outcome measures (symptom questionnaires) should be captured via the study app and the frequency and duration at which these should be delivered via the study app.
Patients with pCF recruited to the PPIE focus groups had previously participated in a separately (MRC) funded study investigating neural mechanisms of pCF. In that study, participants were recruited primarily via social media. Having previously found this a straightforward and useful route for accessing information about Long-COVID/PCS (Post-COVID Syndrome) research and for facilitating their own involvement in research, patients from the PPIE focus groups advocated for a similar approach to recruitment in this study (some members of the PPIE group also participated in interviews for local TV discussing the study, which helped with recruitment).
Feedback from the PPIE survey and focus groups also informed the process of providing participant feedback about the study results. In addition to dissemination through scientific presentation and publication, and via the study website (https://research.ncl.ac.uk/covidfatigue/), the patients also received individualised reports, as had been requested in the feedback from the PPIE surveys. Patient groups were also encouraged to contact the team via the trial website, allowing continued engagement and learning from those significantly affected by pCF.
PAuSing-pCF was a single-site, single-blind, randomised, sham-controlled, interventional trial conducted in adults with post-COVID fatigue. Participants were randomly assigned in equal numbers to one of three treatment groups: active trans-auricular vagus nerve stimulation (taVNS, intervention), sham tragus stimulation (sham, control 1), or active pinna (greater auricular nerve) stimulation (placebo, control 2) for 8 weeks, then all participants crossed over to active taVNS for a further 8 weeks. The trial was designed to demonstrate superiority of the active taVNS arm over both control arms, using a two-sided 5% significance level and ensuring adequate power across comparisons. The trial was carried out in the Henry Wellcome Building, Medical School, Newcastle University, Newcastle upon Tyne, UK.
Participants were members of the general population aged 18–65 who had a verified COVID-19 infection confirmed by polymerase chain reaction (PCR), lateral flow, or antigen testing. Only individuals who were not hospitalised during the acute phase of the illness and who were at least four weeks beyond diagnosis were eligible. Recruitment was carried out via the study website and social media, and all applicants were screened for fatigue symptoms prior to enrolment.13
This trial was prospectively registered at https://www.isrctn.com/ISRCTN18015802. Registered May 12, 2022. [Trial registration number: ISRCTN18015802; Clinical Trial Registry: ISRCTN - The UK Clinical Trial Registry (https://www.isrctn.com/); Dates of Clinical Trial: February 2022 to March 2025.]
Eligible participants who provided informed consent were assigned in equal proportions to the three treatment arms (1:1:1). A full description of the inclusion and exclusion criteria is provided in the Extended Data on Figshare (Section 1, Table S1).17 The allocation sequence was generated independently by the statistical team using block randomisation with randomly varying block sizes of three and six to ensure randomization was balanced across groups.
The allocation sequence was uploaded to an in-house app installed on a central computer at the Henry Wellcome Building, Newcastle University, serving as the central randomisation system (allocation concealment mechanism). A research associate, who was separate from the personnel responsible for enrolling participants and assessing eligibility, implemented the random assignments. The enrolment staff did not have access to the randomisation software, ensuring that the allocation sequence remained concealed.
Participants were blinded to their allocated treatment throughout the study. The research associate responsible for patient randomisation was not blinded to the treatment following randomisation. Trial statisticians were also unblinded for the purpose of data analysis and report preparation.
Participants were required to self-administer electrical stimulation to the external ear to activate nerves running beneath the surface of the skin. A FlexiStim (TensCare, Epsom, UK) transcutaneous electrical nerve stimulation (TENS) device was used for this purpose. These devices are commercially available over the counter, or on-line, without prescription.
To activate the auricular branch of the vagus nerve (taVNS) a clip electrode, manufactured in the lab and connected to the TENS system, was attached to the left tragus. Fabric sleeves, cut to the correct length to cover the two prongs of the ear clip electrode, ensured that the saline used to moisten the sleeves made a good electrical contact with skin overlying the tragus. To activate the greater auricular nerve (C2/C3 nerve roots; placebo control), the clip electrode was attached to the left earlobe/pinna.
The stimulation parameters were set for each participant using the electrical muscle stimulation (EMS) program on the TENS machine and the same parameters were used for tragus and earlobe/pinna stimulation. The stimulation protocol began with a two-minute warm up at a frequency of 6 Hz and pulse width of 200 μs. This was followed by a 60-minute train, which alternated 30 seconds at 25 Hz and 300 μs with 60 seconds at 4 Hz and 200 μs. Finally, there was a 3-minute cool down stimulation protocol of 3 Hz and 200 μs. The stimulus current strength was set just above perceptual threshold to ensure the subject was aware of the stimulation, but also so that the intensity was not uncomfortable or painful.
To ensure participants were blinded, they were only required to power the TENS device ON (stimulator settings were pre-set for each participant and the same settings used for all treatments). A small plastic junction box connecting the TENS cables to the earclip cables contained either a 10 MΩ resistor for active stimulation or a 10 Ω resistor to shunt current. The resistor boxes were indistinguishable for participants. The experimenter was not blinded to the treatment after randomisation.
The three treatments were as follows:
Treatment-1 (active taVNS, intervention): Clip electrode was attached to the tragus and connected to the TENS device to stimulate the auricular branch of the vagus nerve. The junction box connecting the TENS cables to the earclip cables contained a 10 MΩ resistor. Because this is much higher than the electrode resistance, almost all current flowed through the electrode, giving active stimulation.
Treatment-2 (sham control): Clip electrode was also attached to the tragus, but the junction box contained a 10 Ω resistor. Because this was much smaller than the electrode resistance, it shunted almost all of the stimulus current. The auricular nerve was therefore not stimulated, but this condition controlled for mechanical pressure exerted by the tragus clip.
Treatment-3 (placebo control): Clip electrode was attached to the earlobe and connected to the TENS device thus stimulating afferents of the greater auricular nerve (C2/C3 nerve roots). Junction box contained a 10 MΩ resistor for active stimulation. This condition controlled for non-specific effects of electrical stimulation.
The sample size was defined a priori in the study protocol.13 Based on previous studies,18,19 the Fatigue Visual Analogue Scale (F-VAS) outcome showed substantial variability and a clinically meaningful difference between treatment arms. The trial was designed to demonstrate superiority of the active taVNS arm over both control arms, using a two-sided 5% significance level and ensuring adequate power across comparisons.
Allowing for adjustment for baseline fatigue VAS in the primary analysis, and accounting for the observed correlation between baseline and follow-up measurements, a total of 32 participants per arm was required to maintain the planned power of 88%.
There was an enclosed box attached to TENS device equipped with a data logger to monitor device usage. It checked every 10 minutes whether the device was active and recorded the usage every 4 hours, allowing assessment of how often the TENS device was used over time. Participants were expected to utilise the device three times each day, with one cycle running for 60 minutes. However, it was anticipated that participants in all arms might deviate from the prescribed plan, leading to non-adherence. In the study, two pre-specified usage patterns were considered, representing two different subpopulations. These patterns are outlined below:
i. A group of participants who used the device at least 150 minutes a day for at least 75% of days (representing a good-adherence group)
ii. A group of participants who used the device at least 60 minutes a day for at least 50% of days (representing a moderate-adherence or minimum acceptable adherence group)
In the following, brief explanations of the study outcomes and variables of interest are provided. More detailed information is available in the Extended Data on Figshare, Section 2.17
2.7.1 Questionnaire-based primary and secondary outcomes
The primary outcome was the change in fatigue severity, measured using the Fatigue Visual Analogue Scale (F-VAS) questionnaire, from baseline to 8 weeks post-intervention, comparing the active arm with each control arm.
Secondary outcomes included several patient-reported and objective measures. Patient-reported outcomes were collected through questionnaires:
• The Sleep Quality Visual Analogue Scale (SQ-VAS) assessed participants’ subjective perception of sleep quality.
• The Functional Assessment of Chronic Illness Therapy – Fatigue Scale (FACIT-F) evaluated self-reported fatigue and its impact on physical, emotional, and social functioning.
• Quality of Life (QoL) was measured using the EQ-5D-5L, a standardised instrument comprising two components: the EQ-5D descriptive system and the EQ Visual Analogue Scale (EQ-VAS). The descriptive system covers five dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression, while the EQ-VAS records the patient’s overall self-rated health. These two components were analysed separately, as they assess different aspects of health-related quality of life.
• The Fatigue Impact Scale (FIS) evaluated fatigue-related impairment across cognitive, physical, and psychosocial domains, with domain scores combined into an overall score.
• The Generalised Anxiety Disorder Scale (GAD−7) measured the severity of anxiety symptoms, including excessive worry, restlessness, and difficulty controlling worry.
• The Patient Health Questionnaire-2 (PHQ-2) screened for depressive symptoms, assessing the frequency of depressed mood and anhedonia over the past two weeks.
• The Karolinska Sleepiness Scale (KSS) measured participants’ level of sleepiness or alertness at a given moment.
• The Brief Pain Inventory – Short Form (BPI-SF) assessed pain severity.
Questionnaires were collected at baseline and follow-up visits at weeks 1, 2, 4, 8, and 16, and were used for descriptive and inferential analyses.
2.7.2 Adverse events
Overall, it was expected that the stimulation would be well tolerated across all treatments and that no serious adverse events would occur. Any adverse events were recorded during treatment and carefully monitored until they resolved, stabilised, or it was confirmed that they were not related to the study procedures.
2.7.3 Neurophysiological measures
Neurophysiological measures were collected using CE-marked (EU-certified) wearable technology and included sleep regularity, step count, duration of inactivity, and duration of light, moderate, and vigorous activity, as well as heart rate variability. These measures were collected at baseline and follow-up visits at weeks 8, and 16.
2.7.3.1 General electrophysiological methods
The assessment protocols follow our previous study, which compared participants with pCF to age-matched controls.5
Electromyogram (EMG) was recorded through adhesive surface electrodes placed on the skin over the muscle belly. EMG signals were amplified and filtered (band-width 30–2000 Hz; D360 8-Channel Patient Amplifier, Digitimer, Welwyn Garden City, UK) and then digitised with a sampling rate of 5 kHz (CED Micro 1401 with Spike2 software, Cambridge Electronic Design, Cambridge, UK) and stored on a computer for off-line analysis. Where a measurement required a constant contraction, visual feedback of rectified and smoothed EMG activity was provided to the subject via a display of coloured bars on a computer screen, calibrated to the subject’s maximum voluntary contraction (MVC). Participants were then asked to maintain a contraction corresponding to 10% of their individual MVC. Transcranial magnetic stimuli were applied using a figure-of-eight coil through a BiStim 2002 stimulator (Magstim, Whitland, UK). The magnetic coil was held to induce electrical currents that flow perpendicular to the presumed line of the central sulcus in a posterior-anterior direction, with the handle pointing backwards and 45° away from the midline. The hotspot was defined as the region where the largest motor-evoked potential (MEP) in the target muscle could be evoked. To ensure a stable coil position during experiments and across sessions, the site of stimulation was marked using a Brainsight neuronavigation system (Rogue Research Inc., Montréal, Canada), which allows online navigation. A Polaris Vicra camera (Northern Digital Inc., Canada) was used to track the coil relative to the head.
2.7.3.2 Peripheral nerve stimulation
Stimuli to peripheral nerves (0.2 ms pulse width) were delivered using a Digitimer DS7AH isolated, constant current stimulator. The size of the maximal M wave was measured by stimulating the median nerve at the wrist and recording EMG from the abductor pollicis brevis (APB) muscle. Stimulus intensity was set to produce a supra-maximal M wave. Ten stimuli at this intensity were delivered and the highest amplitude M wave was used for subsequent normalisation of TMS recruitment curves.
2.7.3.3 TMS recruitment curve
As a measure of motor cortical excitability, the increase in APB MEP with increasing stimulus intensity was used.5 The active motor threshold (AMT) was defined as the intensity which produced a MEP > 100 μV amplitude in at least 3 out of 6 stimuli, while the participant maintained an active contraction of 10% MVC. The intensity of the stimulation was expressed as a percentage of the maximal stimulator output (MSO). Recruitment curves of increasing intensities in 10% MSO steps were obtained in blocks of ten stimuli per step starting at AMT intensity, until 100% MSO was reached.
Offline analysis measured the size of MEPs from single trials and plotted this versus stimulus intensity. A sigmoid curve could then be fitted to the relationship.
2.7.3.4 Paired-pulse TMS
The hotspot was defined as the region where the largest MEP in the first-dorsal interosseous (1DI) muscle could be evoked with the minimum intensity. Resting motor threshold (RMT) was defined as the minimal stimulus intensity needed to produce a MEP > 100 μV amplitude in at least 3 out of 6 stimuli.
For the test stimulus, TMS intensity was adjusted to elicit MEPs of 1 mV amplitude at rest, or to 120% RMT, whichever was lower. The conditioning stimulus intensity was set at 80% RMT. The conditioning stimulus preceded the test stimulus by 3 or 10 ms and the recorded responses were expressed as a percentage of responses to the test stimulus alone, to measure short-interval intracortical inhibition (SICI) and intracortical facilitation (ICF), respectively.5 Twenty stimuli for each condition were given in a pseudo-random order.
2.7.3.5 Galvanic skin response
The galvanic skin response (GSR) is a measure of the cutaneous resistance or conductivity, which can be quantified by passing electricity through a pair of electrodes. Variation in skin resistance depends on sweat production, which itself is mediated by the sympathetic system5; its habituation may be a relevant measure in assessing cognitive states.20 The GSR was measured by placing two metal plates on the lateral and medial surfaces of the index finger. Five loud sounds (115 dB, C weighting, 500 Hz, 50 ms, 8–8.8 s inter-stimulus interval, chosen randomly from a uniform distribution) were played through loudspeakers placed underneath the subject chair, with the subject at rest. The ratio of the GSR response amplitude following the last stimulus compared to the first was used as a measure of habituation.
2.7.3.6 StartReact effect
This paradigm measures reaction time from EMG in response to a visual cue (visual reaction time, VRT), a visual plus quiet auditory cue (visual-auditory reaction time, VART), and a visual plus loud auditory cue (visual-startle reaction time, VSRT). The acceleration of reaction time between VART and VSRT is termed the StartReact effect and assesses connections from the reticulospinal system.5
A green light-emitting diode (LED) was located ~1 m in front of the subject. Participants were instructed to flex their elbow and clench their fist as quickly as possible after the LED illuminated. EMG was recorded from both the first dorsal interosseous muscle (1DI) and biceps muscle, and reaction time measured as the time from cue to onset of the EMG burst. Three types of trial were randomly interleaved (20 repeats per condition; inter-trial interval 6–6.8 s, interval chosen randomly from a uniform distribution): LED illumination alone (VRT), LED paired with a quiet sound (80 dB, 500 Hz, 50 ms, VART), LED paired with a loud sound (120 dB, 500 Hz, 50 ms, VSRT).
StartReact measurements were performed immediately after the GSR habituation test, ensuring that any overt startle reflex had been habituated by the five loud sounds given in that test. The room lights were dimmed for this test.
Data were analysed offline trial-by-trial using a custom MATLAB program which identified the reaction time as the point where the rectified EMG exceeded the mean + 7 SD of the baseline measured 0–200 ms prior to the stimulus. Every trial was also inspected visually, and erroneous activity onset times (caused, for example, by electrical noise artefacts) were manually corrected. Average VRT, VART and VSRT were calculated for each subject and muscle, together with the amplitude of the StartReact effect, equal to VSRT-VART.
2.7.3.7 Stop-signal reaction time
The stop-signal reaction time (SSRT) task measures the ability to inhibit a response after receiving a GO cue. Participants were asked to respond to a GO cue, but to inhibit their responses if a STOP cue appeared. This can indicate the state of premotor cortical-subcortical areas involved in impulse control and allows for increased levels of inhibition in the motor system to be tested. The hardware used to measure SSRT in this study was a portable device recently developed that uses Bayesian statistical analysis to improve the reliability of the measure.21 The battery-powered device consisted of a plastic box which contained a microcontroller and a LCD screen, as well as one green LED, one red LED and a low compliance press button. Participants initiated a trial by pressing and holding down the response button. They were instructed to release this button as quickly as possible when the green LED illuminated (GO trial; 75% of trials).
In 25% of trials, the red LED illuminated 5, 65, 135 or 195 ms after the green LED (stop trial) and participants were instructed not to release the button in these trials. Trials were presented in three blocks of 64, with a 60 s break in between each block. Each block consisted of 48 GO trials and 16 STOP trials (four for each delay). Using the distribution of reaction times on the GO trials, and the proportion of successfully inhibited responses, the algorithm calculated the SSRT as described in full in our previous work.21
2.7.3.8 Electrocardiogram
A single channel of electrocardiogram (ECG) recording was captured, using a differential recording from left and right shoulder (bandpass 0.3–30 Hz) and stored for offline analysis. The time of each QRS complex was extracted. From these times, the mean heart rate and the pNN50 (a measure of heart rate variability and defined as the proportion of successive intervals which differ by >50 ms) were computed. ECG was captured while participants were engaged in the SSRT test (see above), to ensure consistent behaviour across recordings.
2.7.3.9 Twitch interpolation
Post-viral, immune-mediated disorders of the peripheral nervous system cause failure of signal propagation in nerve, ineffective signal transmission at the neuromuscular junction or reduced signal transmission in muscle fibres. If the same occurs in pCF, this would require a stronger voluntary drive to activate muscles and could give rise to a perception of effortful movements.5 These features were tested before and after a maximal voluntary biceps muscle contraction by supramaximal electrical stimulation of the biceps muscle (twitch interpolation (TI)).22
Subjects sat with their arm and forearm strapped into a dynamometer to measure torque about the elbow. The forearm was held vertically in supination, the upper arm horizontal and the elbow was flexed at a 90° angle. Velcro straps held the wrist, forearm and upper arm in place. Thin stainless-steel plate electrodes (30 x 15 mm) covered in saline-soaked gauze were used for electrical stimulation of the biceps muscle, by taping one electrode over the muscle belly and one over its distal tendon. The individual supramaximal stimulus level was set by increasing the intensity until the twitch response (recorded by the dynamometer) grew no further.
Upon receiving an auditory cue, subjects performed a 3 s long maximal voluntary contraction (MVC) until a STOP tone sounded. Electrical stimulation to the biceps was given during MVC, 2 s after the go cue and at rest, 5 s after the stop cue. This sequence was repeated twice, with a 60 s rest period between GO cues. After another 60s rest, another GO cue then indicated to the subject to make a sustained MVC of up to 90s. If exerted force fell below 60% of the initial maximal level, contraction was terminated early. During this long MVC, the biceps muscle was stimulated every 10s. A final three stimuli (inter-stimulus interval 5 s) were given at rest.
If a subject truly performs a maximal voluntary contraction, a superimposed electrical stimulus should not be capable of generating extra force. The size of any elicited twitch thus measures a central activation deficit. Stimulation of a fatigued muscle at rest after the long contraction produces a smaller twitch than that seen before the sustained MVC, indicating peripheral fatigue.
Central activation before and after fatigue, as well as peripheral fatigue were computed based on elicited twitches according to the calculations in McDonald et al.22
2.7.3.10 Biometric data
Various biometric measurements were collected from participants. These included blood oxygen saturation, tympanic temperature, height, weight and full body composition (TANITA BC-545 N Segmental Body Fat Scale).
The main analysis took place once the final 16-week follow-up visit was complete, and all data queries had been resolved. The statistical analysis plan of the study is available on Figshare.23 The completed CONSORT checklist for this study has also been made available on Figshare.24
2.8.1 Analysis population
Both primary and secondary analyses were performed in the randomised population who completed the study, with the analysis set defined as either intention-to-treat (ITT) or modified ITT (when a minimum amount of post-baseline data is required) according to the analysis objectives.
2.8.2 Handling missing data
Because the primary and secondary questionnaires were collected frequently—especially the primary questionnaire, which was collected every other day throughout the trial—the inferential analyses focused on selected time points: baseline and follow-up visits at weeks 1, 8, and 16. For each patient, data were taken from the closest available date within a 7-day window before each time point if there were no missing values; otherwise, the data were recorded as missing. As a result, missingness was not a major issue for the inferential analyses.
Missing data in the neurophysiological measurements for regression analyses were imputed using the mean value for each measurement.
2.8.3 Descriptive analysis
Baseline characteristics and outcome measures were summarised descriptively by randomised group. Continuous variables were summarised using mean and standard deviation or median and interquartile range, as appropriate. Categorical variables were summarised using frequencies and percentages. No formal statistical testing was conducted for baseline characteristics due to the randomised nature of the study.
2.8.4 Primary outcome analysis
To estimate the treatment effect at 8 weeks, two linear regression models—one for each control group—were fitted using the modified ITT population. Week-8 F-VAS was the outcome, adjusted for baseline F-VAS, with indicators for the sham and placebo groups and taVNS as the reference. Adjusted mean differences, 95% confidence intervals, and p-values were reported.
Given repeated F-VAS measurements over time, linear mixed-effects models with a participant-level random intercept were also fitted using F-VAS data from week 1 and week 8 adjusted for baseline F-VAS, using the ITT population. These models also included time-by-treatment interaction terms. Model assumptions were assessed using plots of residuals versus fitted values, along with normal quantile plots of the standardised residuals and standardised predicted random effects.
In the Adherence section (Section 2.6), we noted that some device non-adherence was expected across all treatment groups. To account for this, we applied the Complier Average Causal Effect (CACE) approach using an instrumental variable two-stage least squares method. Two pre-specified prevalent device usage patterns were considered, corresponding to subpopulations with good and moderate adherence. CACE analyses, using the modified ITT basis, were performed separately for each control group (sham and placebo versus taVNS) within each subpopulation, with adjustment for baseline F-VAS.
2.8.5 Secondary analyses
2.8.5.1 Neurophysiological measures
Neurophysiological outcomes were presented descriptively across groups. Associations between baseline neurophysiological measures and F-VAS at weeks 8 and 16 were separately examined within the taVNS group using multivariable linear regression on the modified ITT population. To address multicollinearity and reduce overfitting, lasso regression models were also fitted, with tuning parameters selected using cross-validation.
2.8.5.2 Questionnaire-based secondary outcomes
Patient-reported outcomes, collected through questionnaires (described in Section 2.7.1), were analysed using linear mixed-effects models for data from weeks 1 and 8, on the ITT basis. The models were adjusted for baseline values and included time-by-treatment interaction terms, with random intercepts for participants. Model assumptions were assessed using plots of residuals versus fitted values, along with normal quantile plots of the standardised residuals and standardised predicted random effects.
2.8.5.3 Duration and dose of taVNS
To investigate the optimal duration and dose of taVNS, participants initially allocated to the control groups received taVNS between weeks 9 and 16, resulting in 8 weeks of intervention, while participants in the intervention group received taVNS for 16 weeks. Using F-VAS data from weeks 1, 8, and 16, a linear mixed-effects model adjusted for baseline F-VAS on the ITT basis was fitted to assess whether a longer intervention duration was associated with additional improvement. This was evaluated by comparing week 16 outcomes between the control and intervention groups.
2.8.5.4 Adverse events
Adverse events and serious adverse events were summarised using appropriate descriptive statistics, such as counts by allocated treatment group.
2.8.6 Statistical software
All statistical analyses were performed using R software (version 4.1.3). Analysis and processing of neurophysiological data were performed using MATLAB (TheMathWorks, Inc., Natick, MA, USA).
All data and materials supporting the results and analyses in this paper are publicly available and have been deposited on Figshare.25
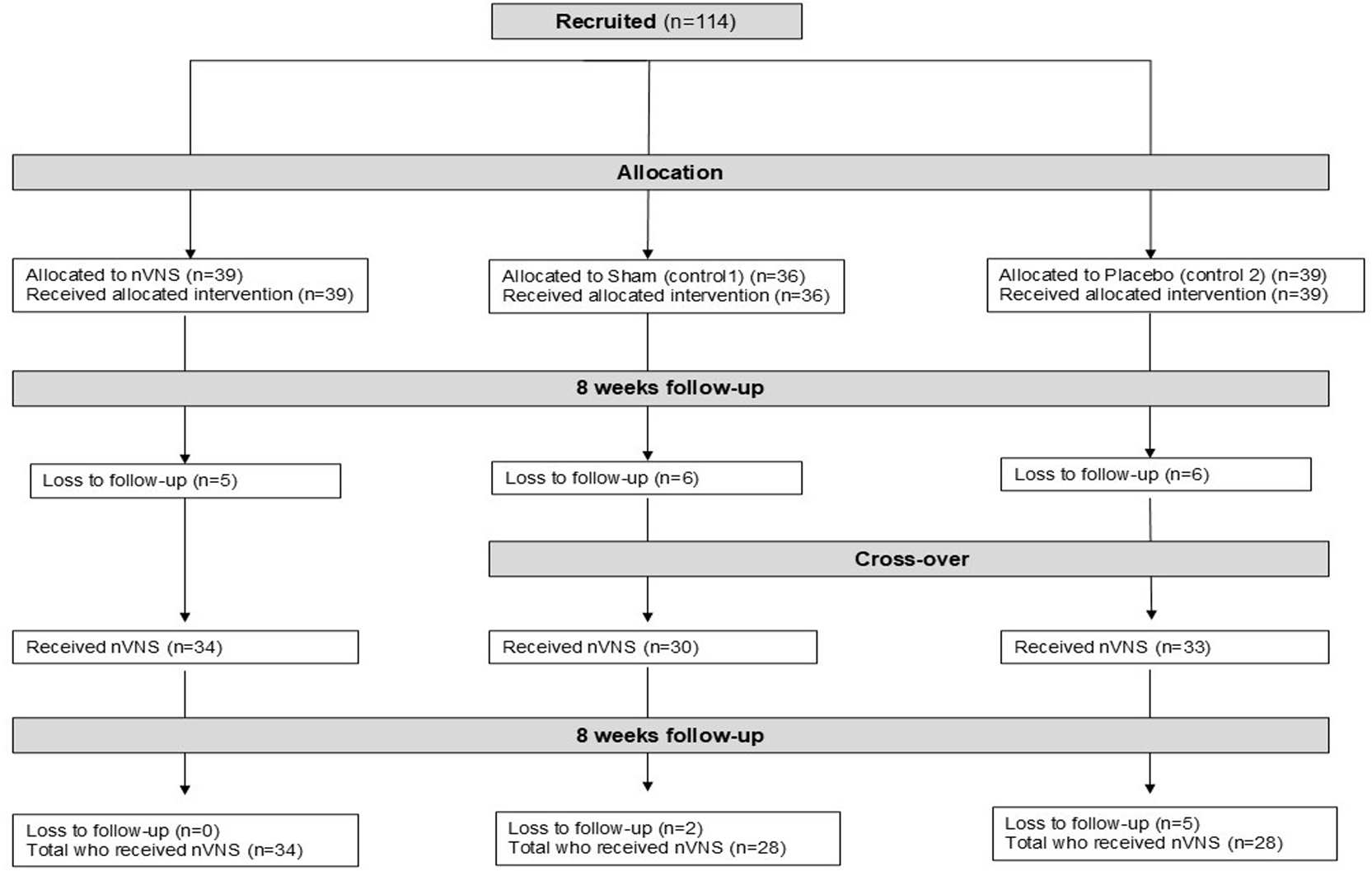
Between May 2022 and July 2024, a total of 114 participants were recruited and randomly assigned in a 1:1:1 ratio to taVNS (n = 39), sham (n = 36), or placebo (n = 39). Seventeen participants were lost to follow-up before the crossover period (week 8), and an additional seven were lost afterwards. Five participants dropped out of the study because of adverse events (see Section 3.3.4). However, the main reason for these losses was that participants simply decided to withdraw from the study for their own personal reasons. The primary analyses were conducted on the 90 participants who completed the trial: 34 in the taVNS group and 28 in each of the sham and placebo groups ( Figure 1). Baseline demographic data were generally balanced across groups ( Table 1). The median age was approximately 50 years, and 74–79% of participants were female. The sham group had a slightly lower median BMI (24 kg/m2) and a shorter median time since COVID-19 (514 days) compared to the other groups.
The primary outcome was the change in F-VAS total score from baseline to week 8. Descriptive analyses indicated a reduction in F-VAS over time in all groups, with a slightly greater decline in the taVNS and placebo groups ( Table 2). Linear regression adjusted for baseline F-VAS showed no statistically significant differences between taVNS and the control groups at week 8 (sham vs taVNS: mean difference 2.6, 95% CI −7 to 12, p = 0.59; placebo vs taVNS: 1.2, 95% CI −9 to 11, p = 0.81; Table 3).
Similarly, a linear mixed model with random intercepts using week 1 and week 8 data found no significant differences (sham vs taVNS: 2.76, 95% CI −5 to 11, p = 0.50; placebo vs taVNS: 2.05, 95% CI −6 to 10, p = 0.62; Table 3).
Model diagnostics for the primary outcome models, demonstrating that the model assumptions were met, are presented in the Extended Data on Figshare (Section 3, Figures S1-S2).17
The proportions of participants classified as good adherence were: 26.67% in the taVNS arm, 13.64% in the sham, and 29.17% in the placebo group. The proportions classified as moderate adherence rates were 76.67% in the taVNS arm, 86.36% in the sham, and 70.83% in the placebo group.
The CACE analysis also did not identify significant effects for either good- or moderate-adherence subgroups ( Table 3).
3.3.1 Neurophysiological measures analysis
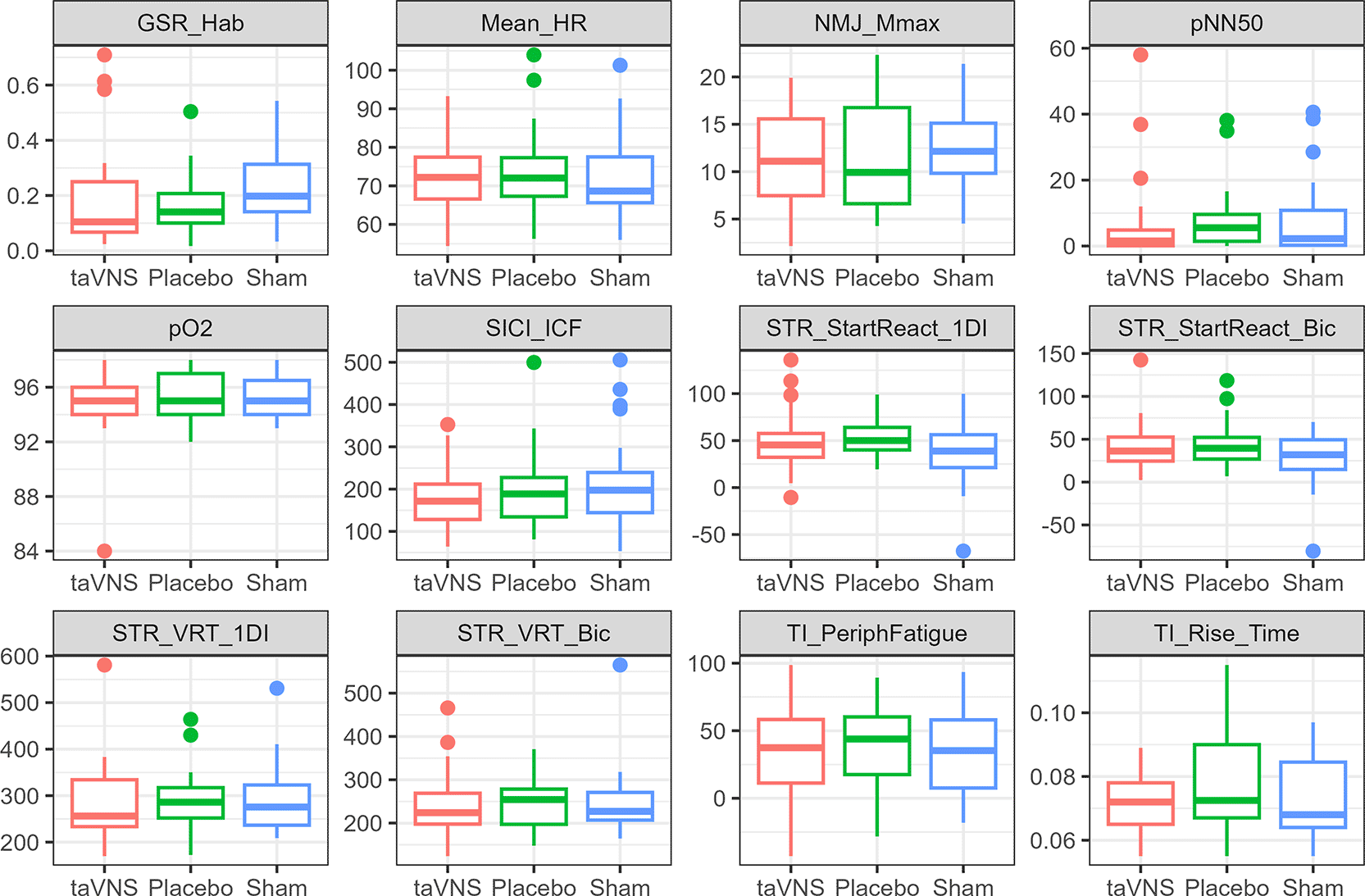
Baseline neurophysiological measurement data, including pO2 (peripheral blood oxygen saturation), NMJ_Mmax (neuromuscular junction, maximal M-wave), SICI_ICF (short-interval intracortical inhibition and intracortical facilitation), GSR_Hab (galvanic skin response habituation), STR_VRT_1DI (StartReact: visual reaction time in 1DI muscle), STR_VRT_Bic (StartReact: visual reaction time in biceps muscle), STR_StartReact_Bic (StartReact: StartReact effect in the biceps muscle), mean_HR (resting heart rate), pNN50 (heart rate variability), TI_PeriphFatigue (twitch interpolation: peripheral fatigue), TI_Rise_Time (twitch interpolation: rise time of muscle EMG), and STR_StartReact_1DI (StartReact: StartReact effect in the 1DI muscle) were considered. The distributions of the neurophysiological measurements across the different groups do not show noticeable differences between groups ( Figure 2). The details of summary statistics for the measurements are available in the Extended Data on Figshare17 (Section 4, Table S2).17
The measurements were also examined as potential predictors of F-VAS in the taVNS group. At week 8, a multivariable linear model identified a significant negative association between STR_StartReact_1DI and F-VAS (p = 0.019), while no other variables were significant. At week 16, pO2, SICI_ICF, STR_StartReact_Bic, TI_PeriphFatigue, and STR_StartReact_1DI were significantly associated with F-VAS ( Table 4). All showed positive associations except STR_StartReact_1DI, which remained negatively associated with F-VAS, consistent with the week 8 findings.
Lasso regression analyses identified pO2, NMJ_Mmax, and TI_PeriphFatigue as significant predictors at week 16, while no significant predictors were identified at week 8 (Extended Data on Figshare, Section 4, Table S3).17
3.3.2 Duration and dose of taVNS analyses
The effect of taVNS duration was assessed by comparing week 16 F-VAS between participants receiving 8 weeks (control groups) versus 16 weeks (taVNS group). No significant differences were observed (sham vs taVNS: −2.90, 95% CI −17 to 11, p = 0.69; placebo vs taVNS: −0.91, 95% CI −15 to 14, p = 0.90; Table 5). Thus there was no significant evidence to suggest that a longer duration of taVNS use had a significantly larger effect compared to a shorter duration.
3.3.3 Questionnaire-based secondary outcome analysis
Fatigue Impact Scale (FIS) scores declined over time across all groups. At week 8, total FIS was significantly higher in the placebo group compared to taVNS (mean difference 9.21, 95% CI 0.7–18, p = 0.03), indicating greater fatigue, while the sham group did not differ from taVNS ( Table 6).
| Adjusted mean difference of total FIS | Standard Error | p-value | 95% confidence interval | |
|---|---|---|---|---|
| Sham vs taVNS | 4.09 | 4.40 | 0.35 | −5 to 13 |
| Placebo vs taVNS | 9.21 | 4.35 | 0.03 | 0.7 to 18 |
For the rest of the patient-reported outcomes, we did not see any significant differences between the taVNS and control groups at week 8. The details of the modelling are available in Table 7 and more details of their descriptive analyses are available in the Extended Data on Figshare, Section 5.17
3.3.4 Adverse events
Overall, the stimulation was well tolerated, though many participants found the earclip to be mildly uncomfortable when it had to be worn on the tragus. No serious adverse events were reported during the study. Three people could not tolerate the pressure from the earclip when having to attach it to the tragus. Two people reported skin irritation at the area of stimulation, which could be treated with emollient cream and resolved after the study. Three people reported nerve pain in their neck, which also radiated to their arms or caused headaches. Only one of these participants was receiving active nVNS at the time, the other two were receiving placebo stimulation to the earlobe. Six participants reported a notable worsening of their fatigue over the course of the first 8 weeks of the study; these divided equally, with two participants receiving each type of stimulation. Out of the 12 people reporting adverse events, 5 decided to drop out of the study as a direct consequence, all of whom believed the stimulation made their fatigue worse and reported no other adverse events. Only one of these participants was receiving active taVNS at the time of drop out.
In this randomised controlled trial, taVNS did not produce a statistically significant reduction in fatigue, as measured by F-VAS, compared with sham or placebo at week 8. Both linear regression and mixed-effects models consistently indicated no significant differences between groups. Similarly, analyses accounting for moderate (minimum)/good adherence (CACE) did not demonstrate meaningful effects.
However, given the low level of adherence across participants, a subgroup analysis which only included the 59 participants who met minimum adherence was conducted.14 In that per-protocol analysis, VAS, FIS scores and peripheral fatigue measured by twitch interpolation improved significantly in the taVNS group. This improvement was absent in the sham or placebo group. These results support taVNS as a potential therapy for pCF mechanistically, but the issue of adherence would have to be addressed for it to be an effective intervention.
Total Fatigue Impact Scale (FIS) scores, as one of the secondary outcomes, revealed modest signals of potential benefit. FIS was lower in the taVNS group compared to placebo at week 8 (with a marginally significant p-value), indicating a possible improvement in fatigue impact, although this was not observed consistently across other secondary outcomes and should be interpreted with caution in light of multiple testing.
Baseline neurophysiological measurements, including pO2, NMJ_Mmax, and TI_PeriphFatigue, were associated with fatigue outcomes at week 16, but not at week 8 based on lasso regression analyses. This suggests that individual neurophysiological differences may influence response to taVNS and warrant further investigation in future mechanistic studies.
Duration of intervention (8 vs 16 weeks) did not significantly modify outcomes, indicating that extending taVNS intervention may not substantially impact fatigue improvement in this study.
These findings highlight the complexity of post-COVID fatigue and suggest that taVNS alone may not be sufficient to produce clinically meaningful changes in the majority of patients. The identification of baseline neurophysiological predictors may inform personalised approaches, enabling targeted selection of patients who might derive greater benefit. Future research should explore combination interventions and consider stratification based on neurophysiological profiles.
This study was approved by the Research Ethics Committee of the Faculty of Medical Sciences, Newcastle University (2284_1/18447/2021). This study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants. This required each participant to sign all study forms, witnessed by an appropriately qualified and delegated member of the research team, before completing any study-specific procedures/investigations.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)