Keywords
Randomised controlled trial, lichen sclerosus, vulval, women’s health, maintenance treatment, topical corticosteroids, economic evaluation, qualitative study
Randomised controlled trial, lichen sclerosus, vulval, women’s health, maintenance treatment, topical corticosteroids, economic evaluation, qualitative study
Vulval Lichen Sclerosus (LS) is a chronic inflammatory condition, with incidence peaks in childhood and post-menopause.1,2 Prevalence is up to 3%, affecting around 1 million women in the UK. Inflammation causes whitening of vulval tissue, bleeding under the skin, texture change and cuts. Patients report itching, pain (particularly during sex) and discomfort in daily activities.2 Paediatric vulval LS accounts for up to 15% of cases.3–5 LS in adults causes progressive loss of vulval architecture (scarring) with vaginal narrowing, burying of the clitoris and resorption of the labia minora. Scarring may occur early in the disease6,7 and is irreversible without surgery.8 LS persisting beyond puberty can prevent normal vulval anatomical development.4 LS also occurs in males, but it is believed to be less prevalent. LS is associated with risk of vulval intraepithelial neoplasia (VIN, a pre-malignant condition) and vulval squamous cell cancer (SCC, an invasive malignancy). Rates of vulval SCC with LS are 20 times higher than in the general population9 with higher mortality than vulval SCC without LS.10
Treatment goals are to control symptoms, prevent scarring and reduce cancer risk. First-line therapy with a very strong topical corticosteroid (TCS) applied daily for three months, combined with vulval care measures11 brings normalisation of skin colour/texture and symptom resolution in 70% of patients.7
However, LS can relapse. Evidence to inform maintenance of remission and prevention of progression is poor. UK guidelines12 are based on expert consensus, a few observational studies and a few low-quality RCTs. A single armed, prospective study of patients with vulval LS in clinical and histological remission suggests 50% will have relapsed after 16 months and 84% after 4 years.13 UK guidelines state that there is no evidence for an optimal maintenance regimen14 and European guidelines reiterate that ‘maintenance’ treatment is a matter of debate.15 A recent commentary noted variation in ongoing management16 and an Australasian consensus statement advises long term individualised proactive maintenance therapy.17 Long term treatment strategy for patients with controlled LS is a research priority area.18
There are no robust long-term RCTs to compare proactive against reactive TCS therapy for LS. An observational study19 suggests symptoms and scarring are reduced by proactive therapy. Another small study (n = 27) reports 60% of patients using only emollients will have a relapse compared with none using proactive TCS over one year.20 Alternative topical treatment options are either unsuitable or ineffective.15–17 Other topical agents on the horizon for LS are still being tested in trials.21,22 TCSs are inexpensive, effective and well understood, and remain the mainstay of LS treatment.
PEARLS will compare TCS maintenance strategies in a superiority trial of proactive over reactive treatment for vulval LS. Paediatric patients are included, due to even greater paucity of evidence for children and young people. There has only been one published randomised control trial with paediatric patients enrolled, this included few children and reported their data together with adult participants, making it difficult to draw any conclusions about the prepubertal patients.23
This study was conducted in accordance with a pre-specified research protocol24 developed by the study team. https://uniofnottm.sharepoint.com/:w:/r/sites/COACHcopy/Shared Documents/General/Post award - Project start-up/TMG/2026/02.67 May 2026/FAMOUS TMG Report v1.0 11May2026.docx?d=w560195273cff414692b96bbe413a9ca7&csf=1&web=1&e=f6lT2l
PEARLS is a two-arm, parallel-group, individually randomised, open label, assessor-blinded, multicentre, superiority trial with a 6-month internal pilot phase. It includes a nested qualitative study and health economic evaluation.
Patients have been involved since the funding application stage, through development of study documents, advising on the trial oversight committees and promoting the study through their networks.
The trial purpose is to compare the clinical and cost effectiveness of a twice weekly topical corticosteroid maintenance strategy (proactive therapy) with as required treatment (reactive therapy) in the management of vulval LS. Study objectives are:
Primary objective.
Compare the effectiveness of proactive versus reactive strategies for using topical corticosteroids on the number of flares within 12 months, in people aged ≥5 years with vulval LS.
Secondary objectives
1. Assess the effectiveness of proactive versus reactive use of TCSs over 24 months in reducing disease progression.
2. Assess the clinical effectiveness of proactive versus reactive strategies for the management of vulval LS for up to 24 months.
3. Assess the safety of using potent and superpotent TCSs in the vulval area over 24 months.
4. Assess the cost-effectiveness of the two treatment strategies.
5. Understand the acceptability of reactive and proactive long-term treatment strategies and the barriers and facilitators to continuing with prescribed treatment.
Participants are recruited across UK specialist primary and approximately 19 secondary care centres. A full list of the research sites can be found at ISRCTN 72275160. Patients were also identified through primary care (as Participant Identification Centres, PICS) and self-referral from the community.
PEARLS is a pragmatic trial and eligibility are designed to fit with usual clinical practice. Diagnosis of LS can be clinical or histological. Diagnosis is confirmed by a clinician with specialist knowledge of vulval skin conditions. Biopsy is not mandated although recommended if there are diagnostic doubt or concerns of malignancy. To enable accurate assessment of progressive anatomical changes, only those born female who have a vulva are eligible for participation. See Figure 1 for eligibility criteria.
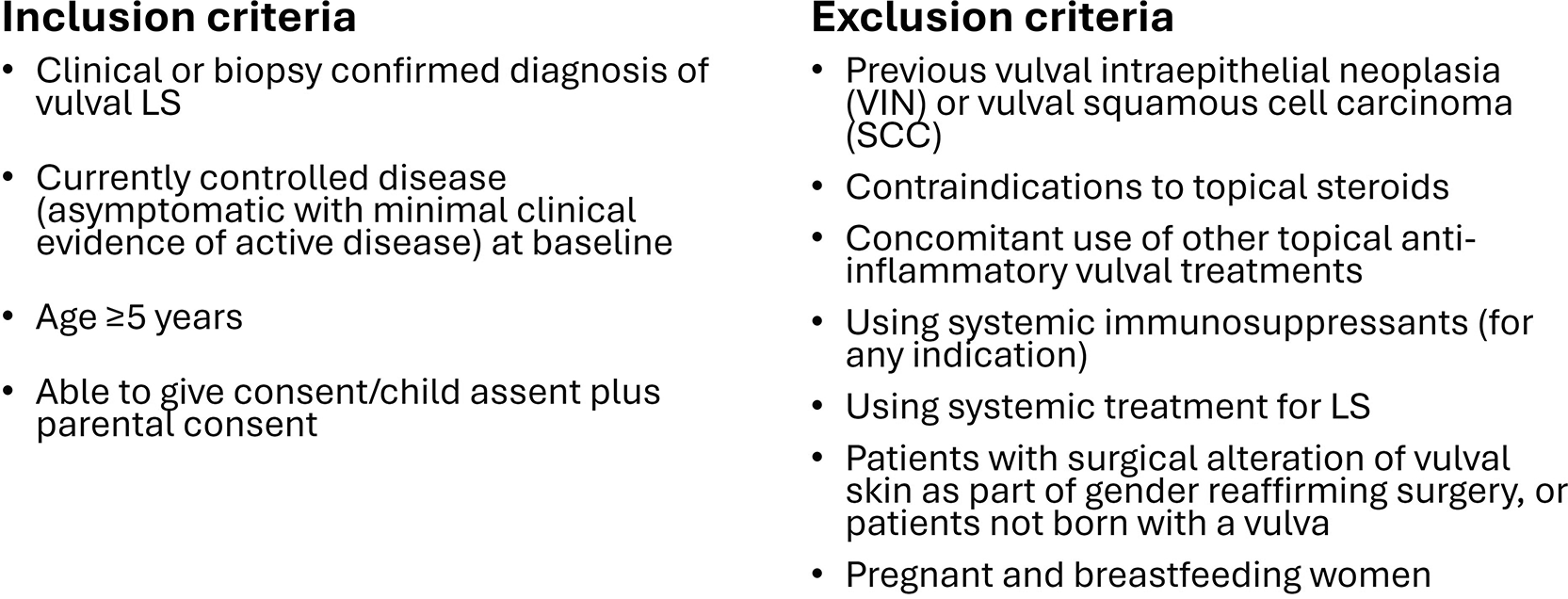
Participants are randomised to one of two treatment strategies:
Intervention group: Potent (strong) or superpotent (very strong) topical corticosteroid (TCS) to be applied on two non-consecutive days per week even in the absence of symptoms (‘proactive’ treatment group).
Comparator group: Potent (strong) or superpotent (very strong) topical corticosteroid (TCS) to be applied as required to treat a LS flare (‘reactive’ treatment group).
As this is a pragmatic trial, participants use the strength of topical corticosteroid recommended by their treating clinician as part of their usual care. If participants experience a flare (regardless of their group allocation) they are instructed to use their TCS once a day for up to one month (up to two weeks for paediatric participants) until their symptoms subside.
If, in the proactive arm, a participant forgets to apply their treatment, they are advised to use their ointment/cream as soon as they remember it, provided there is a gap of at least 24 hours between the doses.
Change of prescribed TCS, or cessation of randomised regimen, is according to usual practice and undertaken at discretion of the managing clinician. This is captured by completion of relevant data in the study database. Reasons to change TCS include difficulty in obtaining specific TCS due to manufacturer issues, or current TCS being ineffective/causing side effects. Reasons to stop randomised regimen include, but are not limited to, frequent flares (if in reactive group) or development of side effects (proactive group).
Concomitant medications only relevant to the condition are collected throughout the study at each study visit.
The trial primary and secondary outcomes are in Table 1.
Given the low-risk nature of the trial, PEARLS has adopted a targeted approach to Adverse Event (AE) and Serious Adverse Event (SAE) reporting.
TCS are established treatments for inflammatory skin disorders, which have a well-documented side effect profile. PEARLS therefore only collects targeted adverse events related to TCS use. Complications of LS with regards to scarring and development of precancerous/cancerous changes are also collected.
SAEs are both defined in the trial protocol and reported, in accordance with UK regulations.
There is no run-in period to the trial except patients who have uncontrolled LS. PEARLS defines controlled disease as patients being asymptomatic with no/mild clinical signs of active disease.
Those screened who do not have well-controlled LS are asked to use their TCS more frequently to gain control before being re-assessed for eligibility. Research visits take place at baseline, 3, 6, 12, 18 and 24 months.
Study recruitment began 01/04/2024 and was initially anticipated to run for approximately 18 months. This was subsequently extended until 31/3/2026.
Sample size calculation was informed using data from a pre-study patient survey. In a subset of 211 survey participants who would be considered eligible, the mean (SD) number of flares was estimated to be 4.0 (2.8) per year. The data showed evidence of overdispersion (variance larger than the mean). Assuming an event rate of 4 flares per patient per year in the control group, a sample size of 320 is required to detect a relative reduction of 25% in number of flares in the intervention group to 3 flares per patient per year, with 90% power, 1:1 allocation and a two-sided significance level of 0.05, using dispersion parameter value of 0.34 estimated from the survey data. A relative reduction in number of symptom flares by 25% is considered meaningful by patients and clinicians since eligible patients have controlled LS, so even a small reduction in number of flares would be meaningful to them. Inflating the sample size by 20% to account for both non-collection of primary outcome data and treatment non-compliance, PEARLS aims to randomise 400 participants.
Trial details have been highlighted through the trial website (www.nottingham.ac.uk/pearls), social media, PPI and charity groups, NIHR’s ‘Be Part of Research’, online health communities and posters in relevant clinical areas. A short animation video is also available on these platforms.
Recruitment of trial participants is through different routes:
Patients and members of the public with lived experience of LS were involved in the development of this study from an early stage. Initial patient and public involvement (PPI) activities took place during the design phase, before the research question and study protocol were finalised. One of the issues PPI activities with the group ‘Patient Advisors for Vulval rEsearch (PAVE)’ highlighted was the long-term impact of LS on quality of life, as well as the burden of recurrent flare-ups, and uncertainty around optimal long-term treatment strategies. Their experiences and priorities directly informed the research question, particularly the comparison between proactive and reactive use of TCSs. PPI contributors emphasised the importance of reducing flare frequency while minimising treatment burden, which shaped the focus of the trial.
PPI contributors are also involved in the ongoing oversight of the study. Patient representatives are regular members of the Trial Management Group and the Trial Steering Committee, contributing to discussions on study conduct, recruitment, implementation, and participants’ experiences throughout the trial. Their input informed key aspects of the study design, including treatment regimens, acceptable frequency of collection of primary and secondary outcome data and follow-up schedules, helping to ensure that study procedures are pragmatic to participants’ lives as well as their current healthcare setting.
Feedback from PPI TMG members and the PAVE group was used to review recruitment materials, including participant information documents, to ensure that language is clear, accessible, and sensitive, supporting appropriate and acceptable recruitment approaches.
Our PPI contributors will be involved in planning the dissemination of study results. The PAVE group will be consulted on how findings are shared with participants and relevant patient communities, including the format and content of lay summaries. Results will be disseminated through patient groups, online platforms, and other channels identified in collaboration with patient representatives.
This trial is managed by the PEARLS Trial Office at the Nottingham Clinical Trials Unit (NCTU), University of Nottingham. Once potential participants have been screened for eligibility and sufficient time allowed to consider their participation, they are asked to complete the informed consent form (ICF) before baseline data is collected by a site investigator to check for further eligibility. If the patient meets all eligibility criteria, they are randomised to the trial treatment.
Trial participation lasts for 24 months, allowing for the assessment of scarring which is considered an important secondary outcome measure and takes longer to occur. Trial assessments are outlined in table 2 and the trial flow chart in Figure 2.
| Trial assessment | Source | Timepoint and visit number | |||||
|---|---|---|---|---|---|---|---|
| Screening/baseline Visit 1 | 3 month Visit 2 | 6 month Visit 3 | 12 month Visit 4 | 18 month Visit 5 | 24 month Visit 6 | ||
| Confirmation of eligibility | Medical notes/clinician/participant | X | |||||
| Consent | Participant | X | |||||
| Randomisation | Participant | X | |||||
| Medical history | Participant/medical notes | X | |||||
| Demographic information | Participant/medical notes | X | |||||
| Concomitant medication (relevant to condition only) | Participant | X | X | X | X | X | X |
| Number of flares and time to 1st flare | Participant | Starting 2 weeks from randomisation, every 2 weeks for 12 months | |||||
| Progression of scarring via examination (clinical diagram), photographs (if consented), and VASS for adults | Clinician | X* | X€¥ | X€¥ | |||
| Vulval development and scarring (adolescents and children) | Clinician | X | X | X | X¥ | X | X¥ |
| Clinical global severity assessment | Clinician | X | X | Xμ | X | Xμ | |
| Condition specific Quality of Life (VQLI for adults or CDLQI for adolescents/children) | Participant | X | X | X | X | X | X |
| Health related quality of life (generic utility instrument) (EQ-5D-5L in adolescents/adults or CHU-9D in children) | Participant | X | X | X | X | X | X |
| Sexual function using Female Sexual Function Index (adults only) | Participant | X | X | ||||
| Acceptability of treatment | Participant | X | X | ||||
| Adherence to treatment | Participant | X | X | X | X | X | |
| Resource use (bespoke and healthcare) | Participant | X | X | X | X | X | X |
| VIN or vulval SCC | Clinician | X | |||||
| Adverse effects of therapy | Participant, Clinician | X | X | X | X | X | |
| Change/cessation of therapy | Participant | X | X | X | X | X | |
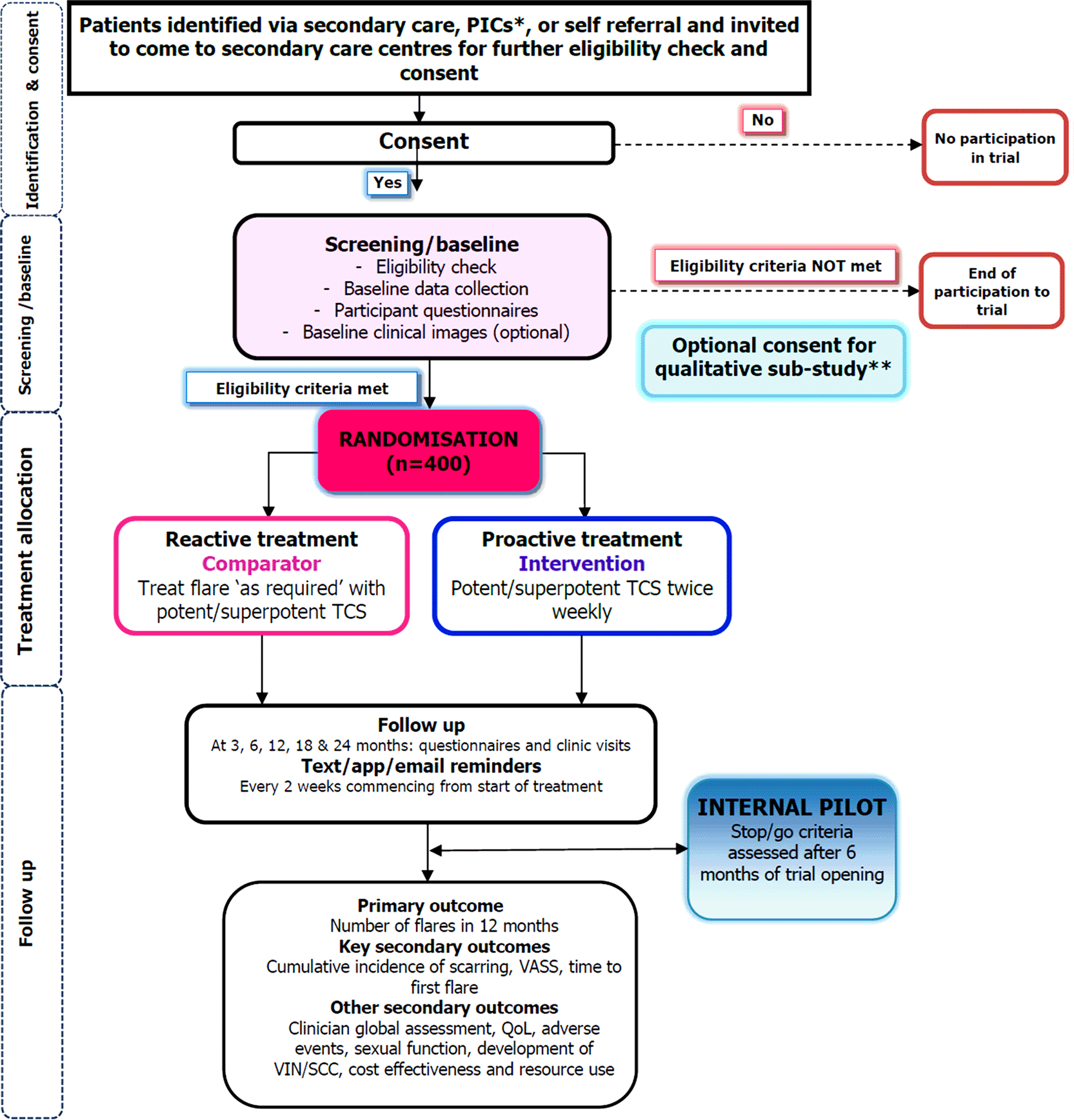
PICs: Participant Identification Centres, SCC: Squamous Cell Carcinoma, TCS: topical corticosteroids, VASS: Vulvar Architecture Severity Scale, VIN: Vulval Intraepithelial Neoplasia.
Randomisation and blinding
Participants are individually allocated in a 1:1 ratio to either proactive or reactive use of TCS. Treatment strategies are assigned randomly using a minimisation algorithm with a random element, balancing across groups on:
• Recruitment by site
• Age at randomisation (prepubertal: 5 to <12 years; adolescent: 12 to <16 years; adult: 16 to <45 years; perimenopausal: 45 years & above)
• Time since last flare (≤6 months, >6 months)
• Strength of study TCS (potent or superpotent)
Trial arm allocation is concealed using a web-based algorithm and held on a secure server at the NCTU. Randomisation is performed by site staff. These personnel do not have access to the random allocation sequence.
Site staff (apart from blinded assessors) are not blinded to allocation due to the nature of the treatment (topical treatment with difference in frequency of use). Details on blinding of other trial staff is listed in table 3.
Blinded assessment of scarring, assessed at 12 and 24 months, by a trained clinician not involved in any other trial procedures, is performed. Global clinical severity is reported by the site’s unblinded clinician at all follow-up time points, but is additionally assessed by the blinded assessor at 12 and 24 months. Revealing the treatment strategy allocation to the assessor should not be necessary as both involve topical steroids. If assessors identify safety concerns, i.e. vulval cancer/VIN, they will report to the unblinded site Principal Investigator (PI) who will inform NCTU.
Clinical photographs are optional and will be taken at baseline (or up to 1 month prior if no active disease during this period), to assess scarring. Additionally, clinicians will document a detailed clinical diagram (using a predefined schematic diagram) for comparison; acting as a backup if photograph quality is insufficient. Subsequent clinical examinations, including blinded assessments of scarring (or failure of normal anatomical development for young people) at 12 and 24 months, will assess changes against the baseline diagram/photographs.
Following successful completion of each research visit, adult participants will receive a £5 high street voucher as inconvenience allowance. All participants can claim up to £15 for expenses incurred in attending their appointment.
Trial data are collected electronically on Research Electronic Data Capture Platform (REDCap) database via electronic Case report Forms (eCRFs).
The primary outcome, collected via a patient flare log, is completed by responding directly to a fortnightly text, email or app notification. The patient response is automatically uploaded to the database.
Other patient reported outcomes (PROs) are completed either electronically, via REDCap survey, or on paper form, depending upon patient preference. Completed paper forms are then transcribed into REDCap at NCTU.
REDCap has built-in validation steps to alert or restrict non-sensical and ambiguous data entry. Any missing and ambiguous data will be queried with the site via e-CRFs. At the end of the trial, prior to data analysis, the data will be validated, and the trial database will be locked to prevent amendment to the data collected. The rules of the data validation and soft/hard database locks are described in PEARLS Data Management Plan (DMP) as part of the internal quality management process.
Participants complete the flare log (primary outcome) every two weeks in response to email, app, or text reminders. Their responses are automatically uploaded to the REDCap database, and database reports are generated to track completion of entries. On occasion, participants who are less confident using certain technology may choose to complete the flare log via a telephone call. In these cases, their responses are recorded and entered into the REDCap database by the research staff on their behalf. Completion of primary outcome is monitored via these reports weekly, as per internal NCTU processes, and those participants who have not responded are contacted by phone/email to obtain the data, where feasible.
Full details on study data management (including participant identifiable information) is provided in PEARLS Data Management Plan.
Trial reporting will be in accordance with Consolidated Standards of Reporting Trials (CONSORT) 2025 guidelines.25 A detailed statistical analysis plan (SAP) will be agreed with the Trial Steering Committee (TSC) prior to database lock. No interim analyses of treatment effectiveness are planned.
Descriptive statistics for the demographic and clinical outcome measures at baseline will be used to assess balance between the randomised arms at baseline. Descriptive statistics appropriate for the outcome will also be presented for all outcomes at all collected time points by treatment arm.
Primary analysis will compare the average number of flares per person-time between the treatment groups, with analysis according to the allocated treatment strategy regardless of adherence to the strategy (intention-to-treat). A random-effects negative binomial regression, adjusting for minimisation factors, with recruitment site adjusted as a random effect and the other factor as fixed effects, and incorporating exposure time (person-time in weeks), will be used to calculate the incident rate ratio (IRR) and the difference in expected flare counts along with the corresponding 95% confidence intervals. Supplementary analyses for the primary outcomes will use per-protocol or complier average causal effect (CACE) analysis if possible, to estimate the effect of the intervention among the participants who would comply with their allocated intervention. Subgroup analyses for the primary outcome will be performed according to age at randomisation (children or adult), time since last flare (≤6 months, >6 months), and strength of prescribed TCS (potent or superpotent), by including appropriate interaction terms in the primary model, however, results will be regarded as exploratory as the trial is not powered to detect interactions.
Between-group comparison of secondary outcomes will be based on an appropriate regression model for the outcome (or appropriate non-parametric estimator), adjusted for the same variables as the primary analysis and baseline outcome values for continuous outcomes if available.
Procedures for missing, unused and spurious data
Complete non-response for primary outcome is expected to be very minimal, as the first data collection timepoint will be within two weeks. The primary negative binomial regression model will account for exposure time (person-time), hence no participant with at least one datapoint will be excluded from the analysis. Should there be other predictors of missingness not adjusted for in the primary model, a sensitivity analysis for effect of missing data will use multiple imputation, based on multivariate imputation by chained equations (MICE), under the missing at random assumption. At least 20 imputations will be performed, and the results combined using Rubin’s rule.
Definition of populations analysed
For the primary analysis, all randomised participants who receive at least one application of the study treatment, will be analysed according to allocated treatment group regardless of adherence to the allocated treatment. This will be supplemented by analysing only the participants who would comply with their allocated intervention. Full definition and cut-off for compliance will be specified in the SAP.
For the secondary outcomes, participants will be analysed according to allocated treatment group regardless of adherence to the allocated treatment.
For the safety outcomes, all randomised participants who receive at least one dose of the study medication will be analysed according to 1) treatment received 2) allocated treatment group regardless of adherence to the allocated treatment.
The trial sponsor is the University of Nottingham. Sponsor form part of essential trial oversight and were involved in the funding and protocol development stages.
PEARLS has a Trial Management Group (TMG) coordinated through the Nottingham Clinical Trials Unit at the University of Nottingham. The TMG meet monthly and they are responsible for oversight of the day-to-day running of the trial.
The Trial Steering Committee (TSC) meets 6-monthly and consists of an independent Chair, other independent members with clinical and research expertise and PPI representative. Members of the TMG and Sponsor representative are non-independent TSC members. The TSC role is to provide overall supervision on behalf of the Sponsor and Funder and to ensure the trial is carried out according to regulatory standards.
The Data Monitoring Committee (DMC) comprises completely independent members who have expertise in LS, statistics and trials. The DMC monitors outcome data, safety data and other trial data to make recommendations for the TSC on whether there are any ethical or safety reasons why the trial should not continue. They meet 6-monthly.
This study has received ethical approval by South West – Central Bristol Research Ethics Committee- Health Research Authority (HRA) (Reference 24/SW/0016 on 7/3/2024). Independent ethical approval was sought through the Health Research Authority (HRA) rather than the affiliated University ethics committee because the study involves human participants recruited through NHS services. In the UK, research involving NHS patients, their data, or NHS sites is required to undergo review by an NHS Research Ethics Committee and HRA approval to ensure compliance with national regulatory, governance, and ethical standards. University ethics approval alone would not have been sufficient for this type of research.
All participants will provide written informed consent before they undergo any study interventions. The Participant Information Sheet (PIS) will be provided to patients in advance, where possible, to ensure that they have sufficient time to consider their participation. Informed consent will be taken at the screening/baseline visit, by the Investigator or delegate appropriately trained to do so. If needed, the hospital interpreter and translator services will be available to help with the understanding of the trial and discuss the PIS/consent forms. Study documentation will only be available in English. The Informed Consent Form will be signed and dated by the participant before they enter the trial and countersigned by the Investigator (or delegate).
Where the participant is under age 16, an appropriate age range PIS will be provided. Parental or legal guardian consent must be obtained, however, in addition to parent/guardian consent, assent will be obtained for all children. The assent will be formally captured (signed) for those 12 years and older. In the event of any conflict between the parent and child, the child WILL NOT enter the study. Once a participant reaches the age of legal consent, adult PIS and a specific re-consent form will be used to re-consent.
Amendments to the protocol are requested by application to the MHRA/REC. These are detailed on the amendment log and once approved, communicated to all participating sites with accompanying updated documentation.
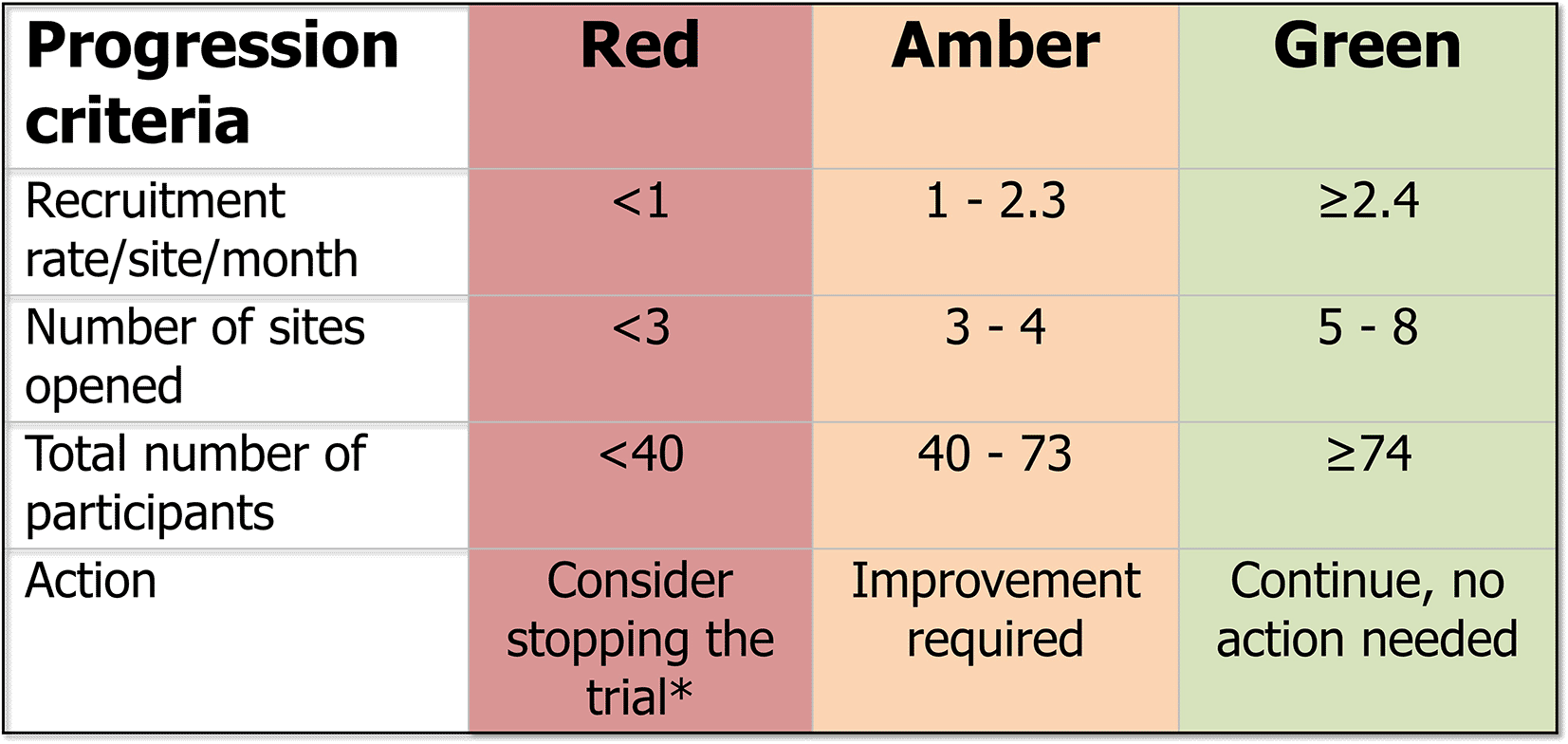
An internal pilot phase is included to monitor the recruitment of sites and participants against agreed milestones ( Figure 3).
A nested qualitative study, led by co-applicants at the University of Bristol, is integrated into PEARLS with the main goals of:
1. Identifying ways to optimise recruitment outcome completion and other trial processes (during pilot phase)
2. Understanding the acceptability of reactive and proactive long-term treatment strategies and the barriers and facilitators to continuing with prescribed treatment (main study phase).
Optional consent is taken at baseline for those willing to be contacted about the qualitative study.
During the internal pilot phase, remote interviews with people who entered or declined PEARLS (or parents and guardians of paediatric patients) and staff involved in recruitment are planned. Qualitative researchers will also observe site initiation visits and drop-in sessions. Observations and interviews will be analysed rapidly using framework analysis and fed back to the TMG. Timely implementation of any potential improvements will then be made to optimise recruitment processes.
The second phase of qualitative interviews takes place when participants reach the 12-month primary endpoint. Interviews with 10–12 adult participants from each trial group will take place for participants to reflect over their experience in the trial and give their thoughts on how they will manage their LS beyond the trial. The experiences of up to 22 children and young people with vulval LS (and their parents/carers) will be explored through a separately funded study called ‘CHILL’ (‘Understanding the lived experiences and unmet needs of CHILdren with Lichen sclerosus and their carers’ NIHR206722). These interviews take place via teleconference, videoconference or face-to-face, depending on participants’ preferences. Patients aged 14–17 may choose to answer the same questions in a written free text online survey, if they do not wish to participate in an interview. Reflexive thematic analysis26,27 will be used to analyse all second phase interviews.
A within trial health economic evaluation will provide evidence on value for money of the specific interventions as well as around the range of costs incurred by people with LS and the validity of utility instruments for this condition. An incremental cost analysis will be conducted from an NHS and Personal Social Services (PSS) perspective in the primary analysis capturing the intervention resource use and wider health resource use related to LS (including primary care, secondary care, emergency care and medications as well as any resource use related to adverse effects) throughout the trial period. A quasi-societal perspective incorporating changes to time at work and personal out-of-pocket costs (e.g. travel to appointments, over the counter products, alternative medicine etc) associated with vulval LS will be reported separately. Resource use will be valued using published unit costs or participant reported estimates for a common recent price year.
Generic utility instruments to measure health-related quality of life will be measured with EQ-5D-5L (adolescents and adults) and Child Health Utility Instrument - Nine Dimensions (CHU-9D) (children – completed by parental proxy for 5–6 year olds and self-completed for all other ages) at baseline, 3, 6, 12, 18 and 24 months and valued using the appropriate value sets at the time of analysis. These values will be used to estimate Quality-Adjusted Life-Years (QALYs) using linear interpolation and area under the curve with and without baseline adjustment.
The primary analysis will be a cost effectiveness analysis over 12 months using the primary clinical outcome, as this will enable all participants to be analysed in one analysis irrespective of age. However, since it is unclear what value decision makers place on these clinical outcomes we will also undertake two cost-utility analysis (one analysis will focuse on adolescents and adults, and where sufficient data exists one for children, reflecting the use of different utility instruments in these two groups) to estimate incremental cost per Quality-Adjusted Life Years. Sensitivity analyses will be undertaken to explore key uncertainties in the economic evaluation, including the impact of any missing data, perspective and timeframe. Where the clinical statistical analysis finds important differences in exploratory sub-group analyses, exploratory subgroup analysis will be undertaken to estimate the cost-effectiveness for these groups also. A health economic analysis plan will be written and reviewed.
Study results will be published in relevant high-impact journals. We will present the results at appropriate academic conferences.
The dissemination strategy and materials will be developed with our PPI contributors and TMG members to ensure findings are conveyed in meaningful ways to patients. We anticipate that infographics and patient story video(s) will be shared on social media. Authorship will be determined according to ICMJE principles and the use of professional writers is not planned.
The PEARLS study will inform the long-term treatment of vulval LS. If evidence of superiority for proactive therapy is found, this will lead to a reduction of flares (thereby improving QOL) and possibly minimise disease progression and reduce cancer development. An important safety message about TCS use in vulval skin will be conveyed to patients and patient groups. Strategies to encourage patients to adhere to long term treatment will be developed through in-depth qualitative work. If proactive therapy is not found to be superior, patients and clinicians can make an informed choice as to which strategy is preferred for the individual.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with NIHR Open Research
Already registered? Sign in
If you are a previous or current NIHR award holder, sign up for information about developments, publishing and publications from NIHR Open Research.
We'll keep you updated on any major new updates to NIHR Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)